336/2004 Sb.
NAŘÍZENÍ VLÁDY
ze dne 5. května 2004,
kterým se stanoví technické požadavky na zdravotnické prostředky a kterým
se mění nařízení vlády
č. 251/2003 Sb., kterým se mění některá nařízení vlády vydaná k provedení
zákona č. 22/1997 Sb.,
o technických požadavcích na výrobky a o změně a doplnění některých
zákonů,
ve znění pozdějších předpisů
Změna: 212/2007 Sb.
Změna: 245/2009 Sb.
Změna: 65/2011 Sb.
Vláda nařizuje podle § 22 zákona č. 22/1997 Sb., o technických požadavcích
na výrobky a o změně a doplnění některých zákonů, ve znění zákona č.
71/2000 Sb. a zákona č. 205/2002 Sb., (dále jen "zákon") k provedení
§ 11 odst. 1 a 2, § 11a, § 12 a 13 zákona a k provedení zákona č. 123/2000 Sb., o
zdravotnických prostředcích a o změně některých souvisejících
zákonů, ve znění zákona č. 130/2003 Sb. a zákona č. 274/2003 Sb., (dále
jen "zákon o zdravotnických prostředcích"):
ČÁST PRVNÍ
TECHNICKÉ POŽADAVKY
NA ZDRAVOTNICKÉ PROSTŘEDKY
Úvodní ustanovení
§ 1
Toto nařízení zapracovává
příslušné předpisy Evropské unie1) a upravuje
a) technické požadavky na zdravotnické prostředky2) a
b) podrobné specifikace, pokud jde o rizika přenosu transmisivních (přenosných)
spongiformních encefalopatií (dále jen "TSE") na nemocné fyzické osoby
(dále jen "pacient") nebo jiné fyzické osoby použitím, za obvyklých
podmínek, zdravotnických prostředků určených ke styku s lidským tělem,
ne však ke styku pouze s neporušenou kůží, vyrobených s využitím neživé
zvířecí tkáně nebo neživých výrobků vyrobených ze zvířecí tkáně v případě,
že tato tkáň pochází ze skotu, ovcí nebo koz, a též z jelenů, norků,
losů a koček; náplň podrobných specifikací je uvedena v příloze č. 12 k tomuto nařízení.
§ 2
(1) Stanovenými výrobky podle § 12 odst. 1 zákona jsou pro účely posuzování
shody podle tohoto nařízení zdravotnické prostředky s výjimkou
a) diagnostických zdravotnických prostředků in vitro3) a
b) aktivních implantabilních zdravotnických prostředků.4)
(2) Toto nařízení se nevztahuje na
a) léčivé přípravky5); při rozhodování o tom, zda výrobek
spadá do oblasti působnosti zákona o léčivech5)
nebo tohoto nařízení vlády, se především
přihlíží k hlavnímu způsobu účinku výrobku,
b) kosmetické přípravky,
c) transplantáty nebo tkáně nebo buňky lidského původu
a výrobky obsahující tkáně nebo buňky lidského
původu nebo z nich odvozené, s výjimkou
zdravotnických prostředků uvedených v § 2 odst. 2 písm. g) zákona o zdravotnických
prostředcích,
d) lidskou krev, výrobky z krve, lidskou krevní plazmu nebo krevní složky
lidského původu nebo zdravotnické prostředky obsahující v době svého
uvedení na trh6) takovéto výrobky z krve, krevní plazmu nebo krevní
buňky, s výjimkou zdravotnických prostředků uvedených v § 2 odst. 2
písm. g) zákona o zdravotnických prostředcích,
e) transplantáty, tkáně nebo buňky zvířecího původu, s výjimkou prostředků
vyrobených s využitím neživé zvířecí tkáně nebo neživých výrobků ze
zvířecí tkáně odvozených.
§ 3
Vymezení pojmů
(1) Pro účely tohoto nařízení, pokud jde o zdravotnické prostředky
uvedené v § 1 písm. b), se rozumí
a) buňkou nejmenší organizovaná jednotka jakékoli živé formy, která
je ve vhodném prostředí schopna nezávislé existence a vlastní obnovy,
b) tkání jakákoli organizace buněk, popřípadě mimobuněčných složek,
c) derivátem materiál získaný výrobním procesem z tkáně zvířat, například
kolagen, želatina, monoklonální protilátky,
d) neživým být bez jakékoli možnosti metabolizovat ani se rozmnožovat,
e) přenosnými původci neklasifikovaní patogenní činitelé, priony a takoví
činitelé, jako jsou původci bovinních spongiformních encefalopatií
(dále jen "BSE") a klusavky (scrapie),
f) snížením, vyloučením nebo odstraněním proces, kterým se sníží počet
přenosných původců nebo kterým se přenosní původci vyloučí, popřípadě
odstraní, aby se zabránilo infekci nebo patologické reakci,
g) inaktivací proces, jímž se snižuje schopnost přenosných původců vyvolat
infekci nebo patologickou reakci,
h) zemí původu země, v níž bylo zvíře narozeno, chováno, popřípadě poraženo,
i) výchozími materiály suroviny nebo jakékoli jiné prostředky zvířecího
původu, z nichž, nebo s jejichž pomocí se vyrábějí zdravotnické prostředky
uvedené v § 1 písm. b).
(2) Uvedením do provozu se rozumí okamžik, ve kterém je zdravotnický
prostředek splňující požadavky tohoto nařízení předán uživateli,8)
u něhož je instalován a připraven poprvé k určenému účelu použití.
§ 4
Obecné zásady
(1) Zdravotnický prostředek musí vyhovovat požadavkům uvedeným v příloze č. 1 k
tomuto nařízení (dále jen "základní požadavky") a požadavkům,
které se na konkrétní prostředek vztahují, a to s přihlédnutím k určenému
účelu použití tohoto prostředku. Existuje-li příslušné riziko, musí zdravotnické
prostředky,
které jsou zároveň strojním zařízením8a), splňovat
rovněž základní požadavky na ochranu zdraví
a bezpečnost stanovené v příloze č. 1 k nařízení vlády
o technických požadavcích na strojní zařízení8a), pokud
jsou tyto základní požadavky na ochranu zdraví a bezpečnost
specifičtější než základní požadavky stanovené
v příloze č. 1 k tomuto nařízení.
(2) Základní požadavky se považují za splněné, jestliže zdravotnický
prostředek je v souladu s požadavky harmonizovaných norem,9) které se k tomuto zdravotnickému
prostředku
vztahují s přihlédnutím k určenému účelu použití.
(3) Na trh nebo do provozu může být zdravotnický prostředek uveden,
jestliže
a) byla u něho stanoveným způsobem posouzena shoda jeho vlastností se
základními požadavky (dále jen "shoda") a výsledkem tohoto posouzení
bylo zjištění, že zdravotnický prostředek základním požadavkům vyhovuje,
je opatřen, s výjimkou zakázkového zdravotnického prostředku a zdravotnického
prostředku určeného ke klinickým zkouškám, označením CE,10)
splňuje další požadavky uvedené v odstavci 1 a výrobce11) nebo zplnomocněný
zástupce12) vydal o tom písemné prohlášení (dále jen "prohlášení o
shodě"), a
b) byly k němu přiloženy informace o jeho použití v souladu s bodem
13 přílohy č. 1 k tomuto nařízení; při jeho uvedení na trh v České
republice musí být informace o jeho použití v českém jazyce.
(4) Zdravotnický prostředek určený k provedení klinické zkoušky13) může být za
tímto účelem předán lékaři nebo jiné osobě, která je
na základě své kvalifikace oprávněna provádět klinické zkoušky, za
podmínek stanovených v zákoně o zdravotnických prostředcích,14) v § 15
a v přílohách č. 8 a 10 k tomuto nařízení; tento prostředek nesmí být
opatřen označením CE.
(5) Zakázkový zdravotnický prostředek15) může
být uveden na trh a do provozu, jestliže u něho byla
posouzena shoda v souladu s § 9 odst. 5 a s přílohou č. 8 k tomuto nařízení. K zakázkovým
zdravotnickým
prostředkům tříd IIa, IIb a III musí být přiloženo prohlášení
uvedené v příloze č. 8 k tomuto nařízení, které
je k dispozici konkrétnímu pacientovi.
(6) V případech, kdy zdravotnický prostředek není v souladu s tímto
nařízením nebo ohrožuje zdraví nebo životy lidí nebo pokud zdravotnický
prostředek nese označení CE a je řádně uveden na trh nebo do provozu
anebo je zdravotnický prostředek řádně udržovaný a používaný v souladu
s určeným účelem použití, a přesto existuje nebezpečí, že může negativně
ovlivnit zdraví lidí, se zřetelem na povahu rizika pro uživatele, se
postupuje podle zákona, zákona o zdravotnických prostředcích a podle
zvláštního právního předpisu.16) O
takových případech a následných opatřeních informuje Ministerstvo průmyslu
a obchodu neprodleně Evropskou komisi a Ministerstvo
zdravotnictví (dále jen "ministerstvo"). Provedená opatření musí být
řádně odůvodněna, zejména musí být uvedeno, zda ochranné opatření bylo
učiněno na základě
a) nedodržení základních požadavků,
b) nesprávného použití harmonizovaných norem podle § 4 odst. 2, pokud
se uvádí, že byly použity, nebo
c) nedostatku v samotných harmonizovaných normách podle § 4 odst. 2.
(7) Rozhodnutí podle zákona, zákona o zdravotnických prostředcích,
zvláštního právního předpisu16) a tohoto nařízení týkající se odmítnutí
nebo omezení při uvádění zdravotnického prostředku na trh nebo do provozu
nebo provádění klinických zkoušek anebo stažení zdravotnických prostředků
z trhu musí obsahovat konkrétní důvody, které k tomu vedly.
(8) Zdravotnický prostředek využívající jadernou energii nebo zdroj
ionizujícího záření musí být posouzen z hlediska radiační ochrany osobou
s odpovídající akreditací pro tuto činnost a s povolením podle zvláštních
právních předpisů.17)
(9) Základní požadavky z hlediska elektromagnetické kompatibility
jsou konkretizovány pro stanovené výrobky podle tohoto nařízení v příloze č. 1 k
tomuto nařízení a při posuzování těchto výrobků se nepoužije
zvláštní právní předpis.18)
(10) Je-li výrobek určen výrobcem k používání
jako osobní ochranný prostředek7) a zároveň jako
zdravotnický prostředek ve smyslu tohoto nařízení,
musí rovněž splňovat příslušné základní požadavky
na ochranu zdraví a bezpečnost podle nařízení vlády,
kterým se stanoví technické požadavky na osobní
ochranné prostředky7).
§ 5
Označování zdravotnických prostředků
(1) Zdravotnické prostředky, které nejsou zakázkové ani určené pro
klinické zkoušky, musí před uvedením na trh nebo do provozu splňovat
požadavky tohoto nařízení a musí být opatřeny označením CE. Pokud je
označení CE zmenšeno nebo zvětšeno, musí být dodrženy vzájemné poměry
rozměrů tohoto označení. Jednotlivé části označení CE musí mít zásadně
stejné vertikální rozměry, které nesmějí být menší než 5 mm; tento
minimální rozměr může být pro zdravotnické prostředky malých rozměrů
upraven.
(2) Pokud zdravotnický prostředek podléhá z jiných hledisek i požadavkům
stanoveným jinými právními předpisy, které stanoví povinnost opatřit
jej označením CE, pak toto označení vyjadřuje, že zdravotnický prostředek
vyhovuje i těmto předpisům. V dokumentaci, upozorněních nebo
návodech, požadovaných příslušnými právními předpisy, a přiložených
k příslušným zdravotnickým výrobkům, musí však být uveden seznam použitých
právních předpisů a dále směrnic, jak byly zveřejněny v Úředním věstníku
Evropské unie, jejichž požadavky byly těmito právními předpisy
převzaty.
(3) Označení CE musí být umístěno viditelně, čitelně a nesmazatelně
na zdravotnickém prostředku nebo jeho sterilním obalu, pokud je to
proveditelné a vhodné, a dále v návodu na použití. Pokud je to možné,
musí být označení CE i na obalu zdravotnického prostředku, ve kterém
se prodává.
(4) K označení CE musí být připojeno, s výjimkou zdravotnického prostředku
třídy I, který není sterilní ani s měřicí funkcí, identifikační číslo
notifikované osoby, která u zdravotnického prostředku posoudila shodu
některým z postupů uvedených v přílohách č. 2, 4, 5 a 6 k tomuto nařízení.
(5) Na zdravotnickém prostředku nesmějí být umístěny značky a popisy,
které by omylem mohly být považovány za znaky významem nebo graficky
se podobající označení CE. Jiný znak lze umístit na zdravotnický prostředek,
jeho obal nebo v návodu doprovázejícím zdravotnický prostředek za předpokladu,
že jím není snížena viditelnost ani čitelnost označení CE.
(6) Jestliže se pro účely předvádění zdravotnického prostředku na
výstavách, veletrzích i jinak použije zdravotnický prostředek, který
neodpovídá požadavkům tohoto nařízení, musí být tento prostředek viditelně
označen tak, aby bylo patrné, že nemůže být uveden na trh a do provozu,
dokud nebude uveden do souladu s tímto nařízením.
§ 6
Neoprávněné připojení označení CE
(1) Jestliže vznikne důvodné podezření, že označení
CE bylo ke zdravotnickému prostředku připojeno
neoprávněně nebo toto označení chybí, postupuje se
podle zvláštního právního předpisu19).
(2) Jestliže výrobce nebo jeho zplnomocněný zástupce nesplní podmínky
pro označení CE a opatření uložená Českou obchodní inspekcí týkající
se zdravotnického prostředku dotčeného podle odstavce 1, postupuje
se podle zvláštních právních předpisů.20)
(3) O opatřeních podle odstavců 1 a 2 informuje
Evropskou komisi a příslušné úřady členských států
Evropské unie nebo Evropského sdružení volného obchodu,
který je současně smluvní stranou Evropského
hospodářského prostoru, (dále jen „členský stát“) Ministerstvo
průmyslu a obchodu.
(4) Ustanovení odstavců 1 až 3 se vztahují i na případy, kdy označení
CE bylo připojeno v souladu s postupy podle tohoto nařízení na výrobky,
na které se toto nařízení nevztahuje.
§ 7
Klasifikace
(1) Zdravotnické prostředky se zařazují podle míry rizika, kterou
představuje jejich použití pro uživatele, popřípadě pro jinou fyzickou
osobu, do tříd I, IIa, IIb a III; zařazování zdravotnického prostředku
do některé z těchto tříd se provádí podle pravidel uvedených v příloze č. 9 k tomuto
nařízení.
(2) Jestliže ministerstvo dospěje k závěru, že
a) použití klasifikačních pravidel podle přílohy č. 9
k tomuto nařízení vyžaduje rozhodnutí o klasifikaci
určitého zdravotnického prostředku nebo
určité kategorie zdravotnických prostředků,
b) určitý zdravotnický prostředek nebo určitá kategorie
zdravotnických prostředků by měly být zařazeny
odchylně od ustanovení přílohy č. 9 k tomuto
nařízení do jiné třídy,
c) shoda zdravotnického prostředku nebo kategorie
zdravotnických prostředků by měla být posouzena
výlučně podle 1 z postupů uvedených v § 9,
d) je potřeba rozhodnout, zda konkrétní výrobek
nebo skupina výrobků vyhovuje některé z definic
v § 2 zákona o zdravotnických prostředcích, nebo
e) klasifikační pravidla stanovená v příloze č. 9 k tomuto
nařízení vyžadují přizpůsobení z důvodu
technického pokroku nebo informací, které se stávají
dostupnými v systému evidence nežádoucích
příhod,
požádá Evropskou komisi, aby učinila
nezbytná opatření; žádost musí být řádně odůvodněna.Postupy posuzování shody
§ 8
(1) Výrobce nebo zplnomocněný zástupce provádí nebo zajišťuje u zdravotnického
prostředku posouzení shody postupy a úkony, které jsou uvedeny v §
9 až 12.
(2) Na pokyn výrobce je zplnomocněný zástupce oprávněn zahájit postup
podle příloh č. 3, 4, 7 a 8 k tomuto nařízení.
§ 9
(1) U zdravotnického prostředku třídy III, který není zakázkový ani
určený pro klinické zkoušky, se výrobce pro jeho opatření označením
CE řídí postupem podle
a) ES prohlášení o shodě podle přílohy č. 2 (Systém úplného zabezpečení
jakosti) k tomuto nařízení, nebo
b) ES přezkoušení typu podle přílohy č. 3 k tomuto nařízení ve spojení
s postupem pro
1. ES ověřování podle přílohy č. 4 k tomuto nařízení, nebo
2. ES prohlášení o shodě podle přílohy č. 5 (Zabezpečení jakosti výroby)
k tomuto nařízení.
(2) U zdravotnického prostředku třídy IIa, který není zakázkový ani
určený pro klinické zkoušky, se výrobce pro jeho opatření označením
CE řídí postupem pro ES prohlášení o shodě podle přílohy č. 7 k tomuto
nařízení ve spojení s postupem pro
a) ES ověřování podle přílohy č. 4 k tomuto nařízení, nebo
b) ES prohlášení o shodě podle přílohy č. 5 (Zabezpečení jakosti výroby)
k tomuto nařízení, nebo
c) ES prohlášení o shodě podle přílohy č. 6 (Zabezpečení jakosti zdravotnického
prostředku) k tomuto nařízení,
nebo podle odstavce 3 písm. a).(3) U zdravotnického prostředku třídy IIb, který není zakázkový ani
určený pro klinické zkoušky, se výrobce pro jeho opatření označením
CE řídí postupem pro
a) ES prohlášení o shodě podle přílohy č. 2 (Systém úplného zabezpečení
jakosti) k tomuto nařízení s výjimkou bodu 4 této přílohy, nebo
b) ES přezkoušení typu podle přílohy č. 3 k tomuto nařízení ve spojení
s postupem pro
1. ES ověřování podle přílohy č. 4 k tomuto nařízení,
2. ES prohlášení o shodě podle přílohy č. 5 (Zabezpečení jakosti výroby)
k tomuto nařízení, nebo
3. ES prohlášení o shodě podle přílohy č. 6 (Zabezpečení jakosti zdravotnického
prostředku) k tomuto nařízení.
(4) U zdravotnického prostředku třídy I, který není zakázkový ani
určený pro klinické zkoušky, se výrobce pro jeho opatření označením
CE řídí postupem podle přílohy č. 7 k tomuto nařízení a vypracuje ES
prohlášení o shodě před uvedením tohoto zdravotnického prostředku na
trh.
(5) Před uvedením každého zakázkového zdravotnického prostředku na
trh výrobce postupuje podle bodů 1., 2.1., 3.1. a 4. přílohy č. 8 (Prohlášení
o zdravotnických prostředcích pro zvláštní účely) k tomuto nařízení
a vypracuje prohlášení stanovené touto přílohou. Výrobce předá České
obchodní inspekci i Státnímu ústavu pro kontrolu léčiv (dále jen "Ústav")
na jejich vyžádání seznam zakázkových zdravotnických prostředků, které
byly uvedeny do provozu v České republice tímto výrobcem.
§ 10
(1) Při posuzování shody u zdravotnického prostředku výrobce, popřípadě
notifikovaná osoba, zohledňuje výsledky hodnocení a ověřovacích postupů,
pokud byly provedeny v souladu s tímto nařízením v jednotlivých mezistupních
výrobního procesu.
(2) Jestliže postup posuzování shody u zdravotnického prostředku zahrnuje
účast notifikované osoby, výrobce nebo jeho zplnomocněný zástupce požádá
o tuto službu podle svého výběru notifikovanou osobu s odpovídajícím
rozsahem autorizace.
(3) Notifikovaná osoba může požádat výrobce o nezbytně nutné informace
pro ověření shody z hlediska zvoleného postupu posuzování shody.
(4) Certifikáty vydané notifikovanou osobou a jiná její rozhodnutí
v souladu s přílohami č. 2, 3, 5 a 6 k tomuto nařízení jsou platné po dobu nejdéle
5 let a mohou být prodlouženy o další období v délce vždy nejvýše 5 let, a to na
základě žádosti výrobce podané v době uvedené ve smlouvě mezi výrobcem a notifikovanou
osobu, která certifikát vydala.
(5) Průvodní a výrobní dokumentaci zdravotnického prostředku, záznamy
a korespondenci týkající se postupů uvedených v § 9 lze pořizovat v
úředním jazyce členského státu, ve kterém jsou
postupy prováděny, popřípadě v jiném jazyce, který je pro notifikovanou
osobu přijatelný.
§ 11
Zvláštní postup pro systémy
a soupravy zdravotnických prostředků a pro
provádění sterilizace zdravotnických prostředků
(1) U systémů a souprav zdravotnických prostředků se postupuje odchylně
od § 9 a 10.
(2) Osoba, která sestavuje zdravotnické prostředky opatřené označením
CE k určeným účelům použití, aby je uvedla na trh jako systém nebo
soupravu, vypracuje prohlášení, ve kterém uvede, že
a) ověřila vzájemnou kompatibilitu sestavených zdravotnických prostředků
podle pokynů jejich výrobců a provedla operace pro provoz podle těchto
pokynů,
b) zabalila systém nebo soupravu zdravotnických prostředků a připojila
k ní odpovídající informace pro uživatele včetně pokynů od výrobců
jednotlivých zdravotnických prostředků a
c) její činnost při sestavování zdravotnických prostředků odpovídá příslušným
metodám vnitřních kontrol a inspekcí.
(3) Nejsou-li splněny podmínky uvedené v odstavci 2, zejména nejsou-li
zdravotnické prostředky tvořící systém nebo soupravu zdravotnických
prostředků opatřeny označením CE nebo není-li systém či souprava zdravotnických
prostředků kompatibilní z hlediska původních určených účelů použití
jednotlivých zdravotnických prostředků, považuje se takový systém nebo
souprava zdravotnických prostředků za samostatný zdravotnický prostředek,
na který se vztahuje postup podle § 9 a 10.
(4) Osoba, která za účelem uvedení na trh sterilizuje soupravy a systémy
zdravotnických prostředků uvedené v odstavci 2 nebo zdravotnické prostředky,
které byly opatřeny označením CE s tím, že jsou jejich výrobci určeny
ke sterilizaci před jejich použitím v rámci poskytování zdravotní péče,
zvolí jeden z postupů uvedených v přílohách č. 2 nebo 5 k tomuto nařízení.
Použití uvedených příloh a činnost notifikované osoby se omezuje na postup
potřebný k dosažení sterility trvající, dokud není sterilní balení otevřeno nebo
poškozeno. Osoba provádějící sterilizaci vypracuje prohlášení, že sterilizace
byla provedena v souladu s pokyny výrobce.
(5) Zdravotnické prostředky uvedené v odstavcích 2 a 4 nesmí být dodatečně
opatřeny označením CE, musí však být k nim přiloženy informace v souladu
s bodem 13 přílohy č. 1 k tomuto nařízení, které obsahují příslušné
údaje výrobců o zdravotnických prostředcích, které byly sestaveny.
Prohlášení podle odstavců 2 a 4 uchovává osoba uvedená
v odstavcích 2 a 4 po dobu nejméně 5 let pro
potřebu příslušných správních úřadů.
§ 12
Postupy posuzování shody z hlediska minimalizace rizika přenosu nákazy
TSE na člověka
(1) Před podáním žádosti o posouzení shody podle § 9 odst. 1 u zdravotnického
prostředku uvedeného v § 1 písm. b) výrobce provede analýzu rizika
a management rizika (dále jen "řízení rizika") podle přílohy č. 12
k tomuto nařízení.
(2) Kolagen, želatina a lůj používané při výrobě zdravotnických prostředků
uvedených v § 1 písm. b) musejí splňovat nejméně takové požadavky,
které by musely splňovat, pokud by byly určeny k lidské spotřebě.
(3) Zdravotnické prostředky uvedené v § 1 písm. b) lze uvádět na trh
a do provozu pouze za předpokladu, že odpovídají specifikacím stanoveným
v příloze č. 12 k tomuto nařízení.
(4) Posuzování shody zdravotnického prostředku uvedeného v § 1 písm. b) zahrnuje
hodnocení splnění základních požadavků a specifikací stanovených
v příloze č. 12 k tomuto nařízení. Notifikované osoby zhodnotí záměr
výrobce při rozboru analýzy rizika a řízení rizika, a to zejména
a) informace poskytnuté výrobcem,
b) odůvodnění použití zvířecích tkání nebo derivátů zvířecího původu,
c) výsledky eliminačních, popřípadě deaktivačních studií nebo rešerše
odborné literatury,
d) výrobcem provedenou kontrolu jeho dodavatelů, výchozích materiálů
a konečných výrobků,
e) nutnost ověřovat původ výchozích materiálů, včetně dodávek od dodavatelů
výrobce.
(5) Notifikované osoby při hodnocení analýzy rizika a řízení rizik
budou v rámci posuzování shody brát ohled na certifikát ověření nepřítomnosti
TSE vydaný Evropským ředitelstvím pro jakost léků (dále jen "certifikát
TSE"), pokud je pro výchozí materiály k dispozici.
(6) S výjimkou zdravotnických prostředků vyrobených za použití výchozích
materiálů, pro které byl vydán certifikát TSE podle odstavce 5, osoby,
které provádějí hodnocení závěrů analýz rizik a řízení rizik neživých
zvířecích tkání nebo derivátů zvířecího původu určených k použití při
výrobě zdravotnických prostředků uvedených v § 1 písm. b), zjišťují
prostřednictvím ministerstva stanoviska příslušných úřadů ostatních
členských států týkající se jejich hodnocení
závěrů analýz rizik a řízení rizik předmětných tkání a derivátů, které
provedli výrobci.
(7) Před vydáním certifikátu ES přezkoumání návrhu zdravotnického
prostředku (dále jen "certifikát ES přezkoumání návrhu") a certifikátu
ES přezkoušení typu zdravotnického prostředku (dále jen "certifikát
ES přezkoušení typu"), notifikovaná osoba přihlédne ke stanoviskům
přijatým v průběhu 12 týdnů od data, kdy byla vyžádána stanoviska příslušných
úřadů členských států.
§ 13
Oznamovací povinnosti
(1) Výrobce, který v souladu s postupy podle § 4 odst. 3 uvádí zdravotnické prostředky
na trh nebo do
provozu, zplnomocněný zástupce nebo osoba, která se
podílí na činnostech uvedených v § 11, oznamuje v elektronické
podobě ministerstvu
a) zahájení činnosti v souladu s přílohou č. 13 k tomuto
nařízení,
b) ukončení činnosti v souladu s přílohou č. 13 k tomuto
nařízení,
c) uvedení zdravotnického prostředku na trh v souladu
s přílohou č. 15 k tomuto nařízení, a to ještě
před uvedením zdravotnického prostředku na trh,
d) ukončení uvádění zdravotnického prostředku na
trh v souladu s přílohou č. 15 k tomuto nařízení a
e) změnu některého z údajů oznámených podle písmen
a) a c).
Údaje podle přílohy č. 15 k tomuto nařízení se oznamují
až poté, co oznamovatel obdrží od ministerstva
evidenční číslo na základě splnění oznamovací povinnosti
v souladu s přílohou č. 13 k tomuto nařízení.
Výrobce zakázkového zdravotnického prostředku
oznamuje v elektronické podobě ministerstvu pouze
údaje podle písmen a) a b) a změnu některého z údajů
oznámených podle písmene a).(2) Osoba, uvádějící zdravotnický prostředek na
trh nebo do provozu na území České republiky, je
povinna na žádost ministerstva poskytnout informace
umožňující identifikaci zdravotnického prostředku
a podklady, které sloužily k posouzení shody.
(3) Jestliže výrobce, který zamýšlí uvést pod svým
jménem na trh zdravotnický prostředek podle odstavce 1 nebo 2, nemá sídlo v členském
státě, pověří
pro uvádění těchto zdravotnických prostředků na trh
zplnomocněného zástupce, který je jeho jediným
zplnomocněným zástupcem v rámci členských států.
(4) Ministerstvo na žádost sdělí členskému státu
a Evropské komisi informace vyplývající z odstavců 1 až 3 poskytnuté výrobcem nebo
jeho zplnomocněným
zástupcem.
(5) Distributor, dovozce a osoba provádějící servis
zdravotnických prostředků oznamují v elektronické
podobě ministerstvu
a) zahájení činnosti v souladu s přílohou č. 14 k tomuto
nařízení,
b) ukončení činnosti v souladu s přílohou č. 14 k tomuto
nařízení a
c) změnu některého z údajů oznámených podle písmene
a).
§ 14
Soubor údajů
(1) Údaje
a) o zdravotnických prostředcích a osobách uvedených
v § 13,
b) o vydaných, změněných, pozastavených a zrušených
certifikátech, jakož i o zamítnutí žádostí o vydání
certifikátu podle postupů stanovených v přílohách č. 2 až 7 k tomuto nařízení,
c) získané v souladu s postupem upravujícím oznamování
a evidenci nežádoucích příhod21) a
d) o klinických zkouškách
se zpracovávají v souladu s tímto nařízením v informačním
systému podle § 41 zákona o zdravotnických prostředcích
do doby, kdy osoba nakládající se zdravotnickými
prostředky oznámí ministerstvu ukončení činnosti
a po dobu dalších 20 let. Údaje z informačního
systému jsou přístupné ministerstvu, Úřadu pro
technickou normalizaci, metrologii a státní zkušebnictví
(dále jen „Úřad“), Ústavu, Státnímu úřadu pro jadernou
bezpečnost u zdrojů ionizujícího záření,
Ústavu zdravotnických informací a statistiky České
republiky a České obchodní inspekci. Údaje z informačního
systému o nežádoucích příhodách22) a klinických
zkouškách zdravotnických prostředků jsou přístupné
pouze ministerstvu, Ústavu a Ústavu zdravotnických
informací a statistiky České republiky
a u zdrojů ionizujícího záření Státnímu úřadu pro
jadernou bezpečnost.(2) Údaje uvedené v odstavci 1 písm. a) a d) se
poskytují v elektronické podobě podle příloh č. 13 až 16 k tomuto nařízení.
§ 15
Klinické zkoušky
(1) U zdravotnických prostředků určených pro
klinické zkoušky výrobce, zplnomocněný zástupce
nebo zadavatel postupuje podle přílohy č. 8 k tomuto
nařízení a oznamuje Ústavu záměr provést klinické
zkoušky formou stanovenou v příloze č. 16 k tomuto
nařízení a současně předkládá prohlášení včetně dokumentace
uvedené v bodě 2.2. přílohy č. 8 k tomuto nařízení.
(2) U zdravotnických prostředků třídy III a implantabilních
zdravotnických prostředků a invazivních
zdravotnických prostředků třídy IIa a IIb s dlouhodobým
použitím lze zahájit klinické zkoušky zdravotnického
prostředku po uplynutí 60 dnů po jejich oznámení
podle odstavce 1, pokud jim Ústav neoznámí
během této lhůty nesouhlas s jejich provedením
z důvodu ochrany veřejného zdraví nebo veřejného
zájmu. Tyto zkoušky lze se souhlasem Ústavu zahájit
i před uplynutím 60denní lhůty, pokud příslušná
etická komise zaujala k programu těchto zkoušek,
včetně přezkoumání plánu klinických zkoušek, kladné
stanovisko.
(3) Klinické zkoušky musí být prováděny v
souladu se zákonem o zdravotnických prostředcích, přílohou č. 10 k
tomuto nařízení a se zvláštními právními předpisy.23)
(4) Ustanovení odstavců 1 a 2 neplatí, jestliže se klinické zkoušky
provádějí za použití zdravotnických prostředků, u nichž byla pro jejich
opatření označením CE posouzena shoda podle § 9, pokud není cílem těchto
zkoušek použití uvedených prostředků pro jiný účel použití, než jim
původně určil výrobce. Příslušná ustanovení přílohy č. 10 k tomuto
nařízení zůstávají nedotčena.
(5) V případě zdravotnických prostředků, které
nejsou uvedeny v odstavci 2, lze klinické zkoušky se
souhlasem Ústavu zahájit ihned po oznámení, a to
za předpokladu, že příslušná etická komise vydala
k programu zkoušek, včetně přezkoumání plánu klinických
zkoušek, kladné stanovisko.
(6) Výrobce, zplnomocněný zástupce nebo zadavatel
oznámí Ústavu ukončení klinické zkoušky, včetně
jeho odůvodnění v případě předčasného ukončení. Je-li
klinická zkouška předčasně ukončena z bezpečnostních
důvodů, předá Ústav toto oznámení všem členským
státům a Evropské komisi. Výrobce nebo zplnomocněný
zástupce uchovává zprávu uvedenou v § 11 písm. c) zákona o zdravotnických prostředcích
k dispozici
příslušným orgánům.
§ 16
Notifikované osoby
(1) Podmínky autorizace jsou stanoveny v příloze č. 11 k tomuto nařízení;
o právnických osobách, které vyhoví požadavkům českých harmonizovaných
norem, které přebírají příslušné harmonizované normy, se předpokládá,
že vyhovují příslušným kritériím pro autorizaci.
(2) Pro provádění postupů posuzování shody pro zdravotnické prostředky
uvedené v § 1 písm. b) musí notifikovaná osoba prokázat aktuální vědomosti
o této problematice, především o podrobných specifikacích stanovených
v příloze č. 12 k tomuto nařízení. V případě zjištění jakýchkoliv nedostatků
v aktuálních vědomostech u notifikované osoby postupuje Úřad podle
zákona.24)
(3) Notifikovaná osoba a výrobce nebo jeho zplnomocněný zástupce na
základě dohody stanoví termíny pro dokončení hodnotících a ověřovacích
činností uvedených v přílohách č. 2 až 6 k tomuto nařízení.
(4) Pokud notifikovaná osoba podle své působnosti zjistí, že výrobce
nesplnil příslušné požadavky tohoto nařízení nebo je nadále neplní,
s výjimkou případů, kdy výrobce zajistil shodu s těmito požadavky zavedením
odpovídajících nápravných opatření, změní nebo zruší certifikát, který
vydala.
(5) Notifikovaná osoba informuje o všech vydaných,
změněných, doplněných, pozastavených a odňatých
certifikátech, jakož i o odmítnutí vydat certifikát,
Úřad a o pozastavených a odňatých
certifikátech a o odmítnutí vydat certifikát informuje
ostatní notifikované osoby. Tyto notifikované osoby
na požádání informuje i o vydaných certifikátech. Notifikovaná
osoba také na požádání zpřístupní veškeré
další související informace.
(6) Při informování příslušných úřadů jiných členských států
a Evropské komise o certifikátech, které notifikovaná osoba změnila nebo
zrušila podle odstavce 4, se postupuje podle zákona.25)
(7) Na žádost poskytne notifikovaná osoba Úřadu všechny příslušné
informace a dokumenty, včetně rozpočtových dokumentů, k ověření plnění
kritérií uvedených v příloze č. 11 k tomuto nařízení.
(8) Úřad poskytne na vyžádání příslušného úřadu jiného členského státu
informace a dokumenty k ověření kritérií uvedených
v příloze č. 11 k tomuto nařízení.
§ 17
Přechodná ustanovení
(1) Platné dokumenty vydané podle § 9 nařízení vlády č. 181/2001 Sb.,
kterým se stanoví technické požadavky na zdravotnické prostředky, ve
znění nařízení vlády č. 336/2001 Sb. a nařízení vlády č. 251/2003 Sb.,
lze využít pro posouzení shody podle tohoto nařízení, pokud nebudou
zrušeny za podmínek stanovených zákonem.
(2) Výsledky zkoušek a zjištění provedených podle § 8 nařízení vlády
č. 181/2001 Sb., kterým se stanoví technické požadavky na zdravotnické
prostředky, ve znění nařízení vlády č. 336/2001 Sb., mohou notifikované
osoby (§ 16) využít jako podklady pro vydání certifikátů a jiných dokumentů
podle tohoto nařízení.
(3) Zdravotnické prostředky, u nichž byla posouzena shoda podle §
8 nařízení vlády č. 181/2001 Sb., kterým se stanoví technické požadavky
na zdravotnické prostředky, ve znění nařízení vlády č. 336/2001 Sb.,
lze uvádět na území České republiky na trh nejdéle do 31. prosince
2005, a to pouze pro uvedení do provozu v České republice.
(4) Zdravotnické prostředky obsahující stabilní deriváty z lidské
krve nebo z lidské plazmy musí být posuzovány ve smyslu bodu 7.4. přílohy č. 1 k
tomuto nařízení. Notifikované osoby přitom zohledňují příslušné
informace týkající se vlastností a funkční způsobilosti těchto prostředků,
zejména výsledky zkoušek a ověřování, provedených dříve podle zákona
č. 79/1997 Sb., o léčivech a o změnách a doplnění některých souvisejících
zákonů, ve znění zákona č. 149/2000 Sb., zákona č. 153/2000 Sb., zákona
č. 258/2000 Sb., zákona č. 102/2001 Sb., zákona č. 138/2002 Sb., zákona
č. 309/2002 Sb., zákona č. 320/2002 Sb., zákona č. 129/2003 Sb. a zákona
č. 274/2003 Sb. Pokud předmětné zdravotnické prostředky vyhovují požadavkům
zákona podle věty druhé, mohou být na území České republiky uváděny
na trh nejdéle do 10. ledna 2007 a do provozu nejdéle do 10. ledna
2009.
(5) Zdravotnické prostředky podle § 1 písm. b), pro které byl vydán
v jiném členském státě Evropských společenství před 1. dubnem 2004
certifikát ES přezkoumání návrhu nebo certifikát ES přezkoušení typu,
popřípadě byl takový certifikát vydán podle § 9 nařízení vlády č. 181/2001
Sb., kterým se stanoví technické požadavky na zdravotnické prostředky,
ve znění nařízení vlády č. 336/2001 Sb. a nařízení vlády č. 251/2003 Sb., lze uvádět
na trh a do provozu nejdéle do 30. září 2004.
Držitel certifikátu ES přezkoumání návrhu nebo certifikátu ES přezkoušení
typu pro předmětný zdravotnický prostředek požádá o dodatek k certifikátu
ES přezkoumání návrhu nebo o dodatek k certifikátu ES přezkoušení typu
prokazující shodu se specifikacemi stanovenými v příloze č. 12 k tomuto
nařízení, pokud bude i nadále uvádět takový prostředek na trh a do
provozu po 30. září 2004.
(6) Zdravotnické prostředky podle § 1 písm. b), u nichž byla posouzena
shoda podle § 8 nařízení vlády č. 181/2001 Sb., kterým se stanoví technické
požadavky na zdravotnické prostředky, ve znění nařízení vlády č. 336/2001 Sb., lze
uvádět na trh a do provozu pouze na území České republiky,
a to nejdéle do 30. září 2004.
(7) Osoby pověřené k činnostem při posuzování shody podle nařízení
vlády č. 181/2001 Sb., kterým se stanoví technické požadavky na zdravotnické
prostředky, ve znění nařízení vlády č. 336/2001 Sb. a nařízení vlády
č. 251/2003 Sb., se považují za pověřené k činnostem podle tohoto nařízení.
(8) Platnost certifikátů notifikovaných osob a jiná jejich rozhodnutí,
která se týkají prsních implantátů, u nichž byla posouzena shoda jako
u zdravotnických prostředků třídy IIb, nelze prodloužit.
ČÁST DRUHÁ
Změna nařízení vlády, kterým se mění
některá nařízení vlády vydaná k provedení zákona
č. 22/1997 Sb., o technických požadavcích na
výrobky a o změně a doplnění některých zákonů,
ve znění pozdějších předpisů
Předseda vlády:
PhDr. Špidla v. r.
Ministr zdravotnictví:
MUDr. Kubinyi, Ph. D. v. r.
Příl.1
ZÁKLADNÍ POŽADAVKY
I.
VŠEOBECNÉ POŽADAVKY
1. Zdravotnické prostředky musí být navrženy a vyrobeny takovým způsobem, aby při
použití za stanovených
podmínek a pro určený účel neohrozily klinický stav nebo bezpečnost pacientů ani
bezpečnost
a zdraví uživatelů, případně dalších osob, a to za předpokladu, že veškerá rizika,
která mohou s určeným
použitím těchto prostředků souviset, jsou přijatelná v porovnání s jejich přínosem
pro pacienta a odpovídají
vysoké úrovni ochrany zdraví a bezpečnosti. To zahrnuje:
1.1. snížení, pokud je to možné, rizika chyby při používání v důsledku ergonomických
vlastností
zdravotnického prostředku a prostředí, ve kterém může být zdravotnický prostředek
používán,
(návrh pro bezpečnost pacientů) a
1.2. zvážení technických znalostí, zkušeností, vzdělání a proškolení, a případně
zdravotního a fyzického
stavu uživatelů, pro které je zdravotnický prostředek určen, (návrh pro laické uživatele,
profesionální
uživatele, postižené osoby nebo jiné uživatele).
2. Řešení, která výrobce zvolí při návrhu a konstrukci
zdravotnických prostředků, musí být v souladu se zásadami
bezpečnosti a se současnou úrovní vědy a techniky. Při výběru
nejvhodnějších řešení musí výrobce vycházet z následujících
zásad v uvedeném pořadí:
2.1. vyloučit nebo minimalizovat veškerá rizika (bezpečným
návrhem a konstrukcí) zdravotnických prostředků,
2.2. učinit, kde je to vhodné, odpovídající ochranná opatření
zahrnující v případě potřeby i varování vůči nebezpečím,
která nelze vyloučit,
2.3. informovat uživatele o přetrvávání rizik v důsledku
nedosažení plné dokonalosti uskutečněných ochranných
opatření.
3. Zdravotnické prostředky musí dosahovat účinnosti určené
výrobcem a být navrženy, vyrobeny a zabaleny tak, aby byly
vhodné pro jednu nebo více funkcí uvedených v § 2 odst. 1
zákona o zdravotnických prostředcích v souladu se specifikací
jejich výrobce.
4. Při zatížení zdravotnických prostředků, které může nastat za
normálních provozních podmínek, nesmí dojít k nepříznivému
ovlivnění jejich charakteristik a účinnosti ve smyslu bodů 1,
2 a 3 do té míry, kterou by došlo k ohrožení klinických stavů
nebo bezpečnosti pacientů, popřípadě jiných osob, a to po dobu
životnosti zdravotnických prostředků uvedenou výrobcem, pokud
výrobce tuto dobu stanovil.
5. Zdravotnické prostředky musí být navrženy, vyrobeny a zabaleny
tak, aby za podmínek stanovených výrobcem pro jejich skladování
a dopravu (například teplota, vlhkost) nemohly být nepříznivě
ovlivněny jejich vlastnosti a účinnost.
6. Jakékoliv vedlejší účinky zdravotnických prostředků musí
představovat pouze přijatelné riziko ve srovnání s jejich
předpokládanými účinky.
6a. Prokázání shody se základními požadavky musí obsahovat klinické hodnocení podle
přílohy č. 10
k tomuto nařízení.
II.
POŽADAVKY NA NÁVRH A KONSTRUKCI ZDRAVOTNICKÝCH PROSTŘEDKŮ
7.
Chemické, fyzikální a biologické vlastnosti zdravotnického
prostředku.
7.1. Zdravotnické prostředky musí být navrženy a vyrobeny tak,
aby byly zaručeny charakteristiky a účinnost uvedené ve
všeobecných požadavcích. Zvláštní pozornost musí být
zaměřena na
7.1.1. výběr materiálů určených pro výrobu a balení
zdravotnických prostředků, zejména z hlediska
toxicity, popřípadě i hořlavosti,
7.1.2. vzájemnou kompatibilitu mezi použitými materiály a biologickými tkáněmi, buňkami
a tělesnými
tekutinami se zřetelem na určený účel použití,
7.1.3. výsledky biofyzikálního nebo modelového výzkumu, jejichž platnost byla již
dříve prokázána.
7.2. Zdravotnické prostředky musí být navrženy, vyrobeny
a zabaleny tak, aby bylo na nejnižší možnou míru sníženo
riziko vyplývající ze znečištění nežádoucími látkami
a složkami záření a jejich reziduí vůči uživatelům
a osobám podílejícím se na dopravě, skladování a používání
zdravotnických prostředků v souladu s určeným účelem
použití. Zvláštní pozornost musí být věnována tkáním
vystaveným působení uvedených nežádoucích látek, složek
záření a jejich reziduí, době trvání a četnosti tohoto
působení.
7.3. Zdravotnické prostředky musí být navrženy, vyrobeny
a zabaleny tak, aby mohly být bezpečně použity společně
s látkami a plyny, s nimiž přicházejí do styku při
normálním použití a obvyklých postupech. Jsou-li
zdravotnické prostředky určeny k podávání léčiv, musí být
navrženy a vyrobeny tak, aby při určeném účelu použití
byly s těmito léčivy kompatibilní v mezích ustanovení
a omezení, kterými se tyto výrobky řídí a aby byla
zachována jejich účinnost v souladu s určeným účelem
použití.
7.4. Ustanovení pro zdravotnické prostředky s integrovaným léčivem nebo derivátem
z lidské krve.
7.4.1. Pokud zdravotnický prostředek obsahuje jako svou integrální součást látku,
která může být
při samostatném použití považována za léčivý přípravek podle zvláštního právního
předpisu5)
a která může působit na tělo doplňujícím účinkem k účinku zdravotnického prostředku,
musí být jakost, bezpečnost a užitečnost této látky ověřena analogicky za použití
metod uvedených ve zvláštním právním předpisu5).
7.4.2. U látek uvedených v bodě 7.4.1. si notifikovaná osoba po ověření užitečnosti
látky jako
součásti zdravotnického prostředku a s přihlédnutím k určenému účelu použití zdravotnického
prostředku vyžádá odborné stanovisko k jakosti a bezpečnosti této látky včetně klinicky
ověřeného rizika při začlenění dané látky do zdravotnického prostředku od jednoho
z příslušných úřadů členských států nebo od Evropské agentury
pro léčivé přípravky (European Medicines Agency), (dále jen „EMEA“). Je-li o odborné
stanovisko požádán Ústav, přihlíží k výrobnímu postupu a k údajům o užitečnosti začlenění
látky do zdravotnického prostředku, které uvedla notifikovaná osoba.
7.4.3. Pokud zdravotnický prostředek obsahuje jako svou integrální součást derivát
z lidské krve,
vyžádá si notifikovaná osoba po ověření užitečnosti tohoto derivátu z lidské krve
jako
součásti zdravotnického prostředku a s přihlédnutím k určenému účelu zdravotnického
prostředku odborné stanovisko EMEA, k jakosti a bezpečnosti tohoto derivátu z lidské
krve, včetně klinicky ověřeného rizika začlenění derivátu z lidské krve do zdravotnického
prostředku.
7.4.4. Jsou-li prováděny změny na doplňující látce začleněné do zdravotnického prostředku,
zejména pokud jde o její výrobní postup, musí být o změnách informována notifikovaná
osoba, která požádá o odborné stanovisko odpovídající příslušný úřad pro léčivé přípravky
(tj. úřad, který vydal původní odborné stanovisko), aby bylo potvrzeno, že jakost
a bezpečnost doplňující látky je zachována. Je-li o odborné stanovisko požádán Ústav,
přihlíží k údajům o užitečnosti začlenění doplňující látky do zdravotnického prostředku,
které uvedla notifikovaná osoba za účelem zajištění, že změny nemají žádný negativní
dopad na stanovené riziko při začlenění této doplňující látky do zdravotnického prostředku.
7.4.5. Jestliže Ústav, jako odpovídající příslušný úřad, který vydal původní odborné
stanovisko,
obdrží informaci o doplňující látce, která by mohla mít vliv na stanovené riziko
při začlenění
doplňující látky do zdravotnického prostředku, sdělí notifikované osobě odborné stanovisko,
zda informace má dopad na stanovené riziko při začlenění doplňující látky do zdravotnického
prostředku či nikoli. Notifikovaná osoba vezme v úvahu aktualizované odborné
stanovisko při přehodnocování svého závěru z postupu posouzení shody.
7.5. Zdravotnické prostředky musí být navrženy a vyrobeny tak, aby se rizika způsobená
látkami
unikajícími ze zdravotnických prostředků snížila na minimum. Zvláštní pozornost je
věnována
látkám karcinogenním, mutagenním nebo toxickým pro reprodukci ve smyslu zákona o
chemických
látkách25a).
Pokud části zdravotnického prostředku nebo zdravotnické prostředky jako celek, které
jsou určené
pro podávání nebo odstraňování léčivých přípravků, tělních tekutin nebo jiných látek
do těla nebo
z těla, nebo zdravotnické prostředky určené pro dopravu a skladování těchto tělních
tekutin nebo
jiných látek, obsahují ftaláty klasifikované jako karcinogenní, mutagenní nebo toxické
pro reprodukci
kategorie 1 nebo 2 ve smyslu zákona o chemických látkách25a), musí být takové zdravotnické
prostředky označeny na samotném zdravotnickém prostředku nebo na obalu každé jednotky
nebo,
je-li to vhodné, na prodejním obalu jako zdravotnický prostředek obsahující ftaláty.
Pokud určené použití těchto zdravotnických prostředků zahrnuje léčbu dětí nebo těhotných
či
kojících žen, musí výrobce pro použití takových látek podat v rámci technické dokumentace
zvláštní odůvodnění s ohledem na shodu se základními požadavky, zejména podle tohoto
bodu,
a v návodech k použití poskytnout informaci o zbytkových rizicích pro uvedené skupiny
pacientů,
a je-li to možné, též o vhodných preventivních opatřeních.
7.6. Zdravotnické prostředky musí být navrženy a vyrobeny tak,
aby rizika nežádoucího vniknutí látek do nich, s ohledem
na zdravotnický prostředek a povahu prostředí, ve kterém
má být použit, byla snížena na nejnižší možnou míru.
8.
Infekce a mikrobiální kontaminace.
8.1. Zdravotnické prostředky a výrobní postupy musí být
navrženy tak, aby se pokud možno vyloučilo nebo snížilo na
nejnižší možnou míru riziko přenosu infekce zdravotnickým
prostředkem na uživatele a jiné fyzické osoby nebo
kontaminace zdravotnického prostředku uvedenými osobami.
8.2. Tkáně zvířecího původu používané k výrobě zdravotnických
prostředků musí pocházet ze zvířat, nad nimiž byl
vykonáván veterinární dozor v rozsahu odpovídajícím
určenému účelu použití těchto tkání.
8.2.1. Informace o geografickém původu těchto zvířat
uchovávají notifikované osoby.
8.2.2. Zpracování, uchovávání, zkoušení a zacházení
s tkáněmi, buňkami a látkami zvířecího původu musí
být prováděno tak, aby bylo dosaženo optimální
úrovně bezpečnosti, zejména vůči kontaminaci viry
při výrobě zdravotnických prostředků, a to
zavedením validovaných metod určených pro
odstraňování nebo inaktivace virů.
8.3. Zdravotnické prostředky dodávané ve sterilním stavu musí
být navrženy, vyrobeny a zabaleny v obalu pro jedno
použití, popřípadě musí být vhodnými postupy zajištěno, že
při uvedení na trh budou sterilní a za stanovených
podmínek skladování a dopravy zůstanou sterilní, dokud
nebude ochranný obal otevřen nebo poškozen.
8.4. Zdravotnické prostředky dodávané ve sterilním stavu musí
být vyrobeny a sterilizovány odpovídajícím schváleným
postupem.
8.5. Zdravotnické prostředky, které mají být sterilizovány,
musí být vyrobeny v příslušně kontrolovaných podmínkách
(například v podmínkách prostředí).
8.6. Obalové systémy nesterilních zdravotnických prostředků
musí zabezpečovat stanovenou úroveň čistoty zdravotnického
prostředku. Jestliže mají být zdravotnické prostředky před
použitím sterilizovány, musí obalové systémy snižovat
riziko mikrobiologické kontaminace na nejnižší možnou
míru. Obalové systémy musí být vhodné pro použití
sterilizační metody stanovené výrobcem.
8.7. Identické nebo podobné zdravotnické prostředky, které jsou
prodávány ve sterilním i v nesterilním stavu, musí být
vzájemně rozlišeny obalem nebo označením.
9.
Konstrukce a vlastnosti zdravotnického prostředku ve vztahu
k prostředí.
9.1. Je-li zdravotnický prostředek určen k použití ve spojení
s jiným zdravotnickým prostředkem nebo příslušným
vybavením, musí být takto vzniklá souprava včetně
propojovacího systému bezpečná a nesmí narušovat
stanovenou účinnost zdravotnických prostředků. Jakékoliv
omezení použitelnosti musí být uvedeno v označení
zdravotnického prostředku (etiketě) nebo v návodu
k použití.
9.2. Zdravotnické prostředky musí být navrženy a vyrobeny tak,
aby byla odstraněna nebo na nejnižší možnou míru snížena
rizika
9.2.1. poranění vyplývající z fyzikálních charakteristik
zdravotnických prostředků, včetně poměru objemu
a tlaku, rozměrových, popřípadě i ergonomických
charakteristik,
9.2.2. spojená se zdůvodněně předvídatelnými podmínkami
okolního prostředí, zejména magnetickým polem,
vnějšími elektrickými vlivy, elektrostatickými
výboji, tlakem, teplotou nebo změnami v tlaku
a zrychlení,
9.2.3. vzájemného ovlivňování s jinými zdravotnickými
prostředky běžně používanými při určitém
vyšetřování nebo terapii,
9.2.4. vyplývající ze stárnutí použitých materiálů nebo
ztráty přesnosti měřicího, popřípadě kontrolního
mechanismu i ze skutečnosti, že zdravotnické
prostředky nelze udržovat nebo kalibrovat
(implantáty).
9.3. Zdravotnické prostředky musí být navrženy a vyrobeny tak,
aby byla odstraněna nebo na nejnižší možnou míru snížena
rizika požáru nebo výbuchu během normálního použití a za
stavu jedné závady. Zvláštní pozornost je nutno věnovat
zdravotnickým prostředkům, které jsou určeny pro použití
i v prostředí hořlavých látek nebo látek, které mohou
vyvolat hoření.
10.
Zdravotnické prostředky s měřicí funkcí.
10.1. Zdravotnické prostředky s měřicí funkcí musí být
navrženy a vyrobeny tak, aby poskytovaly dostatečnou
přesnost a stabilitu v daných mezích přesnosti
s ohledem na jejich určený účel použití; meze přesnosti
stanoví výrobce.
10.2. Stupnice měřidel a displeje musí být řešeny v souladu
s ergonomickými zásadami s ohledem na určený účel
použití.
10.3. Výsledky měření provedených zdravotnickými prostředky
s měřicí funkcí musí být vyjádřeny v zákonných
jednotkách podle zvláštních právních předpisů.27)
11.
Ochrana před zářením.
11.1. Obecný požadavek.
11.1.1. Zdravotnické prostředky musí být navrženy
a vyrobeny tak, aby vystavení uživatelů a jiných
fyzických osob účinkům záření bylo s ohledem na
určený účel použití sníženo na nejnižší možnou
míru, aniž by tím bylo omezeno použití
potřebných úrovní záření pro diagnostické
a terapeutické účely.
11.2. Žádoucí záření.
11.2.1. Pokud jsou zdravotnické prostředky určeny
k emitování záření v nebezpečných úrovních, avšak
nezbytných pro specifický zdravotnický účel,
jehož přínos se považuje za odpovídající tomuto
riziku, musí mít uživatel možnost kontrolovat
úroveň těchto emisí. Takové zdravotnické
prostředky musí být navrženy a vyrobeny tak, aby
byla zaručena reprodukovatelnost a tolerance
příslušných proměnných parametrů.
11.2.2. Jestliže jsou zdravotnické prostředky určeny
k emitování potenciálně nebezpečného viditelného,
popřípadě neviditelného záření, musí být
zdravotnické prostředky tam, kde je to možné,
opatřeny displeji, popřípadě doplněné zvukovými
výstrahami, které upozorňují na tyto emise.
11.3. Nežádoucí záření.
11.3.1. Zdravotnické prostředky musí být navrženy
a vyrobeny tak, aby vystavení uživatelů a jiných
fyzických osob nežádoucímu, neužitečnému nebo
rozptýlenému záření bylo omezeno na nejnižší
možnou míru.
11.4. Návod k použití.
11.4.1. Návod, popřípadě provozní instrukce k použití
zdravotnického prostředku emitujícího záření musí
obsahovat podrobné informace o povaze emitovaného
záření, o prostředcích k ochraně uživatele
a o způsobech zamezení zneužití tohoto záření
a vyloučení rizik plynoucích z instalace takového
zdravotnického prostředku.
11.5. Ionizující záření28).
11.5.1. Zdravotnické prostředky určené k emitování
ionizujícího záření musí být navrženy a vyrobeny
tak, aby tam, kde to umožňuje určený účel
použití, bylo možné měnit a kontrolovat množství,
geometrii a jakost emitovaného záření.
11.5.2. Zdravotnické prostředky emitující ionizující
záření určené pro radiodiagnostiku musí být
navrženy a vyrobeny tak, aby příslušné jakosti
zobrazení anebo výstupu pro určený medicínský
účel bylo dosaženo při nejmenší možné radiační
zátěži uživatele těchto prostředků.
11.5.3. Zdravotnické prostředky emitující ionizující
záření určené pro radioterapii musí být navrženy
a vyrobeny tak, aby bylo možné spolehlivé
monitorování a řízení dodávané dávky, typu
a energie svazku záření, a kde je to potřebné,
i jakosti záření.
12.
Požadavky na zdravotnické prostředky připojené ke zdroji
energie nebo vybavené zdrojem energie.
12.1. Zdravotnické prostředky obsahující elektronické
programovatelné systémy musí být navrženy tak, aby byla
zajištěna funkční stálost, spolehlivost a účinnost
těchto systémů v souladu s určeným účelem použití. Při
výskytu závady v tomto systému musí být vhodným způsobem
odstraněna nebo snížena následná rizika na nejnižší
možnou míru.
12.1a. U zdravotnických prostředků, které obsahují programové vybavení nebo které
jsou samy o sobě
zdravotnickým programovým vybavením, musí být programové vybavení validováno podle
nejnovějších
poznatků s přihlédnutím k zásadám vývoje životního cyklu, řízení rizika, validace
a ověřování.
12.2. Zdravotnické prostředky, u nichž závisí bezpečnost
pacienta na vnitřním zdroji energie, musí být vybaveny
zařízením umožňujícím určit stav zdroje energie.
12.3. Zdravotnické prostředky, u nichž závisí bezpečnost
pacienta na vnějším zdroji energie, musí být vybaveny
varovným systémem k signalizaci výpadku tohoto zdroje.
12.4. Zdravotnické prostředky určené k monitorování jednoho
nebo více klinických údajů pacienta musí být vybaveny
odpovídajícími varovnými systémy, které ohlásí
poskytovateli vznik situace, která by mohla vést
k úmrtí pacienta nebo k závažnému zhoršení jeho
zdravotního stavu.
12.5. Zdravotnické prostředky musí být navrženy a vyrobeny
tak, aby bylo na nejnižší možnou míru sníženo riziko
vzniku elektromagnetických polí, která by mohla narušit
provoz jiných zdravotnických prostředků nebo zařízení
v jejich obvyklém prostředí.
12.6. Ochrana před nebezpečím úrazu elektrickým proudem.
Zdravotnické prostředky musí být navrženy a vyrobeny
tak, aby za předpokladu správné instalace a používání
bylo pokud možno vyloučeno nebezpečí náhodného úrazu
elektrickým proudem, a to i za stavu jedné závady.
12.7. Ochrana před nebezpečím vyplývajícím z mechanických
a tepelných vlivů.
12.7.1. Zdravotnické prostředky musí být navrženy
a vyrobeny tak, aby byla zaručena ochrana
uživatele před mechanickým nebezpečím
souvisejícím zejména s pevností, stabilitou
a pohybem některých částí.
12.7.2. Zdravotnické prostředky musí být navrženy
a vyrobeny tak, aby bylo, s přihlédnutím
k současné úrovni vědy a techniky a dostupné
možnosti k omezení vibrací u jejich zdroje,
sníženo na nejnižší možnou míru nebezpečí
vyplývající z vibrací vyvolaných těmito
prostředky, pokud tyto vibrace nejsou
specifickou součástí určeného účelu použití.
12.7.3. Zdravotnické prostředky musí být navrženy
a vyrobeny tak, aby bylo, s přihlédnutím
k současné úrovni vědy a techniky a dostupné
možnosti k omezení hluku zejména u jeho zdroje,
sníženo na nejnižší možnou míru nebezpečí
vyplývající z jimi emitovaného hluku, pokud
tento hluk není specifickou součástí určené
účinnosti zdravotnických prostředků.
12.7.4. Zdravotnické prostředky musí být navrženy
a vyrobeny tak, aby byla snížena na nejnižší
možnou míru rizika vyplývající z koncových
a připojovacích částí těchto prostředků ke
zdrojům elektrické energie, tlakové kapaliny,
vzduchu a plynu, se kterými musí uživatel
zacházet.
12.7.5. Zdravotnické prostředky musí být navrženy
a vyrobeny tak, aby jejich přístupné části
a jejich okolí, s výjimkou částí nebo míst
určených k dodávání tepla nebo k dosažení
stanovených teplot, nedosahovaly za normálních
provozních podmínek potenciálně nebezpečných
teplot.
12.8. Ochrana pacienta před nebezpečím vyplývajícím
z dodávaných energií nebo látek.
12.8.1. Zdravotnické prostředky určené k dodávání energie
nebo látek pacientovi musí být navrženy
a vyrobeny tak, aby dodávané množství mohlo být
regulováno s přesností, která zaručuje bezpečnost
uživatele.
12.8.2. Zdravotnické prostředky určené k dodávání energie
nebo látek pacientovi musí být vybaveny
zařízením, které indikuje, popřípadě zabraňuje
dodávání nepřiměřeného množství energie nebo
látek, které by mohlo být
nebezpečné.Zdravotnické prostředky musí být
vybaveny vhodným zařízením, které je schopné
v nejvyšší možné míře zabránit náhodnému uvolnění
nebezpečných množství energie, popřípadě látky
z jejich zdrojů.
12.9. Na zdravotnických prostředcích musí být zřetelně uvedeny
funkce ovládacích prvků a indikátorů.
Jestliže je na zdravotnickém prostředku umístěn návod
potřebný k jeho provozu nebo uvádějící provozní anebo
nastavovací parametry pomocí vizuálního systému, musí být
tyto informace srozumitelné poskytovateli a podle potřeby
i pacientovi.
13.
Informace poskytované výrobcem.
13.1. Každý zdravotnický prostředek musí být
opatřen informacemi potřebnými pro jeho bezpečné
a správné použití, s přihlédnutím k proškolení a znalostem
potenciálních uživatelů, a k identifikaci výrobce. Těmito
informacemi se rozumějí údaje na etiketě, popřípadě
štítku zdravotnického prostředku a v návodu k jeho
používání; pokud je to proveditelné a vhodné, musí být
tyto informace uvedeny na samotném zdravotnickém
prostředku, na obalu každého jeho kusu, popřípadě na
obalu, ve kterém se prodává. Není-li kusové balení
proveditelné, musí být informace, které jsou uvedené
v první větě tohoto bodu obsaženy v návodu k použití
dodaném s jedním nebo více zdravotnickými prostředky.
V balení zdravotnického prostředku musí být přiložen
návod k jeho použití; výjimečně tato podmínka nemusí být
splněna u zdravotnického prostředku třídy I nebo IIa,
jestliže není pro jeho bezpečné používání nezbytně nutné
dalších informací.
13.2. Jestliže je vhodné, aby informace uvedené v bodu 13.1.
byly v podobě symbolů, musí použitý symbol (značka) nebo
identifikační barva vyhovovat harmonizovaným normám.
V oblastech, pro které harmonizované normy neexistují,
musí být symboly a barvy popsány v dokumentaci dodávané
se zdravotnickým prostředkem.
13.3. Etiketa nebo štítek zdravotnického prostředku musí
obsahovat, zejména
13.3.1. jméno, popřípadě jména a příjmení výrobce, adresu jeho místa podnikání, jestliže
výrobcem
je fyzická osoba; název nebo obchodní firmu, adresu sídla, jestliže výrobcem je
právnická osoba; u zdravotnických prostředků dovážených do Evropské unie s předpokladem
jejich distribuce v rámci Evropské unie musí etiketa nebo štítek, vnější obal nebo
návod k použití navíc obsahovat jméno, popřípadě jména a příjmení nebo název nebo
obchodní firmu a adresu místa podnikání nebo sídla zplnomocněného zástupce, jestliže
výrobce nemá sídlo v členském státě,
13.3.2. podrobné údaje, jež uživatel nezbytně potřebuje k identifikaci zdravotnického
prostředku
a obsahu balení,
13.3.3. podle potřeby nápis "STERILNÍ",
13.3.4. v případě potřeby číslo výrobní dávky (šarže),
před kterým je uvedeno slovo "LOT", nebo sériové
číslo,
13.3.5. rok a měsíc, do kterého lze tento prostředek
bezpečně použít, jestliže přichází tyto údaje
v úvahu,
13.3.6. případně označení, že se jedná o zdravotnický prostředek pro jednorázové
použití.
Informace výrobce o tom, že se jedná o zdravotnický prostředek pro jednorázové použití,
musí být v rámci Evropské unie jednotná,
13.3.7. nápis "Zakázkový prostředek", jde-li o zakázkový
zdravotnický prostředek,
13.3.8. nápis "Pouze pro klinické zkoušky", jestliže je
zdravotnický prostředek určen pro tento účel,
13.3.9. zvláštní podmínky jeho skladování, popřípadě
zacházení s ním,
13.3.10. zvláštní provozní pokyny,
13.3.11. výstrahy, popřípadě i jiná nutná opatření,
13.3.12. rok výroby, jde-li o aktivní zdravotnický
prostředek, pokud se u něho neuvádí obsah bodu
13.3.5.; tento údaj může být součástí čísla
výrobní dávky (šarže) nebo sériového čísla,
pokud je z těchto údajů snadno odvoditelný,
13.3.13. postup při sterilizaci tohoto prostředku,
jestliže přichází v úvahu,
13.3.14. označení, že zdravotnický prostředek obsahuje
derivát z lidské krve, jestliže jde
o zdravotnický prostředek uvedený ve zvláštním
právním předpisu.29)
13.4. Jestliže lze důvodně předpokládat, že určený účel použití
zdravotnického prostředku nemusí být uživateli zřejmý,
výrobce jej uvede v označení zdravotnického prostředku
a v návodu k jeho použití.
13.5. Pokud je účelné a proveditelné, musí být zdravotnický
prostředek a jeho odnímatelné součásti označeny, zejména
údaji o výrobní dávce nebo šarži, aby při určeném účelu
použití bylo možné zjistit rizika představovaná
zdravotnickými prostředky a jejich součástmi.
13.6. Návod k použití, není-li to povahou zdravotnického
prostředku vyloučeno, musí obsahovat:
13.6.1. podrobné údaje uvedené v bodu 13.3., s výjimkou
13.3.4. a bodu 13.3.5. této přílohy,
13.6.2. údaje o účinnosti zdravotnického prostředku podle
bodu 3 této přílohy, jakož i vedlejší nežádoucí
účinky,
13.6.3. podrobné údaje o charakteristikách zdravotnického
prostředku, aby bylo možné určit správné
zdravotnické prostředky nebo vybavení, jejichž
použitím vznikne bezpečný systém nebo souprava,
a to v případě, jde-li o zdravotnické prostředky,
které podle určeného účelu použití musí být
instalovány nebo spojeny s dalšími zdravotnickými
prostředky nebo vybavením.
13.6.4. informace potřebné pro ověření, zda zdravotnický
prostředek je řádně instalován a může být správně
a bezpečně provozován, včetně potřebných údajů
o povaze a četnosti údržby a kalibraci, které
jsou nezbytné k řádné a bezpečné funkci,
13.6.5. informace o možnostech odvrácení nebezpečí
souvisejících s implantací zdravotnického
prostředku jestliže je to potřebné,
13.6.6. informace týkající se nebezpečí vzájemné
ovlivňovatelnosti mezi zdravotnickými prostředky
přítomnými během specifických vyšetření nebo
terapie,
13.6.7. pokyny nezbytné pro případ poškození sterilního
obalu, popřípadě údaje o vhodných postupech při
opětovné sterilizaci,
13.6.8. informace o vhodných postupech, které umožňují
opakované použití zdravotnického prostředku, a to
včetně čištění, dezinfekce, balení, popřípadě
vhodných postupech opětovné sterilizace
zdravotnického prostředku, a doporučovaném počtu
opakovaných použití, jestliže je zdravotnický
prostředek určen k opakovanému použití.
Jestliže je zdravotnický prostředek dodán s tím,
že má být před použitím sterilizován, musí být
návod na čištění a sterilizaci takový, aby při
jeho dodržení zdravotnický prostředek trvale
vyhovoval požadavkům uvedeným v části I. této
přílohy (Všeobecné požadavky). Jestliže je zdravotnický
prostředek označen jako zdravotnický prostředek
pro jednorázové použití, musí návod obsahovat informaci
o známých vlastnostech a technických faktorech zdravotnického
prostředku, které jsou výrobci známy a jež
by mohly při opakovaném použití zdravotnického prostředku
představovat riziko. Pokud podle bodu 13.1.
není potřebný návod k použití, musí být tato informace
dostupná uživateli na požádání,
13.6.9. údaje o zacházení se zdravotnickým prostředkem
před jeho vlastním použitím, zejména
o sterilizaci, popřípadě o konečné sestavě
zdravotnických prostředků,
13.6.10. údaje o povaze, typu, intenzitě a rozložení
emitujícího záření u zdravotnických prostředků
emitujících záření pro zdravotnické účely,
13.6.11. podrobné údaje, které umožní poskytovateli poučit
pacienta o kontraindikacích a nutných předběžných
opatřeních, která je nutno provést v případě změn
účinnosti zdravotnického prostředku,
13.6.12. opatrnost, kterou je nutno za předvídatelných
podmínek prostředí dodržovat, a to zejména
s ohledem na účinky magnetických polí, vnějších
elektrických vlivů, elektrostatických výbojů,
tlaků nebo změn tlaku, zrychlení, zdrojů
tepelného vzplanutí,
13.6.13. přiměřené informace o léčivu, výrobcích,
popřípadě látkách, které se prostřednictvím
zdravotnického prostředku aplikují, včetně
omezení výběru látek, které mají být podávány,
13.6.14. informace týkající se opatrnosti, opatření nutná
pro bezpečné zneškodňování zdravotnických
prostředků, zejména pro případy neobvyklých rizik
souvisejících s touto činností.
13.6.15. informaci o léčivých látkách nebo derivátech z lidské krve obsažených ve
zdravotnickém
prostředku jako jeho integrální součást v souladu s bodem 7.4.,
13.6.16. stupeň přesnosti vyžadovaný u zdravotnických
prostředků s měřicí funkcí,
13.6.17. datum vydání nebo poslední revize návodu k použití.
Příl.2
ES PROHLÁŠENÍ O SHODĚ
(Systém úplného zabezpečení jakosti)
1.
Výrobce zajišťuje zavedení a provozování systému jakosti schváleného pro návrh,
výrobu a výstupní
kontrolu příslušných zdravotnických prostředků podle bodu 3.; systém jakosti podléhá
auditu podle
bodů 3.3. a 4. a podléhá dozoru podle bodu 5.
2.
ES prohlášení o shodě (Systém úplného zabezpečení jakosti) je postup, jímž výrobce,
který plní povinnosti
podle bodu 1., zajišťuje a prohlašuje, že příslušné zdravotnické prostředky splňují
ustanovení
tohoto nařízení, jež se na ně vztahují.
Výrobce
2.1. opatřuje zdravotnické prostředky označením CE v souladu s § 5 a
2.2. vypracuje písemné prohlášení o shodě, které je povinen uchovávat; toto prohlášení
se vypracuje pro
1 nebo více vyrobených zdravotnických prostředků jasně identifikovaných názvem zdravotnického
prostředku, kódem zdravotnického prostředku nebo jiným jednoznačným odkazem.
3.
Systém jakosti.
3.1. Výrobce předkládá písemnou žádost notifikované osobě
o posouzení (vyhodnocení a schválení) jeho systému
jakosti; žádost musí obsahovat
3.1.1. jméno, popřípadě jména a příjmení výrobce, adresu
jeho bydliště a místa podnikání, jestliže výrobcem
je fyzická osoba žijící v zahraničí, jméno,
popřípadě jména a příjmení, adresu trvalého pobytu
výrobce, jestliže má trvalý pobyt v České
republice; obchodní firmu nebo název a adresu
sídla, jde-li o právnickou osobu; v obou případech
včetně adres výrobních míst, pro která platí systém
jakosti,
3.1.2. odpovídající informace o zdravotnickém prostředku
nebo o kategorii zdravotnických prostředků, pro
které postup platí,
3.1.3. písemné prohlášení, že u jiné notifikované osoby
nebyla podána další žádost pro tentýž systém
jakosti vztahující se ke stejnému zdravotnickému
prostředku,
3.1.4. dokumentaci systému jakosti,
3.1.5. záruku výrobce plnit závazky vyplývající ze
schváleného systému jakosti,
3.1.6. záruku výrobce udržovat systém jakosti
v přiměřeném a účinném stavu,
3.1.7. závazek výrobce, že zavede a bude aktualizovat systematický postup vyhodnocování
zkušeností
získaných s vyrobenými zdravotnickými prostředky, včetně ustanovení uvedených
v příloze č. 10 k tomuto nařízení, a vhodným způsobem zavádět nezbytná nápravná
opatření; součástí tohoto závazku je povinnost výrobce oznámit Ústavu nežádoucí příhody
podle zákona o zdravotnických prostředcích30), jakmile se o nich dozví.
3.2. Uplatňovaný systém jakosti musí zajistit, že zdravotnické
prostředky odpovídají ustanovením tohoto nařízení, a to
v každém stadiu od jejich návrhu až po jejich výstupní
kontrolu.
Prvky, požadavky a opatření učiněná výrobcem pro jím
uplatňovaný systém jakosti musí být systematicky a řádně
dokumentovány formou písemně vypracovaných programů
a postupů, jako programy jakosti, plány jakosti, příručky
jakosti a záznamy o jakosti.
To zahrnuje odpovídající dokumentaci, údaje a záznamy
vzniklé na základě postupů uvedených v bodě
3.2.3.
Dokumentace systému jakosti musí obsahovat, zejména popis
3.2.1. cílů jakosti výrobce,
3.2.2. organizace provozovny, zejména
3.2.2.1. organizačních struktur, zodpovědnosti
vedoucích zaměstnanců a jejich pravomocí
ve vztahu k jakosti návrhu a zhotovení
zdravotnického prostředku,
3.2.2.2. metod sledování účinnosti systému jakosti,
především jeho schopnosti dosáhnout
požadované jakosti návrhu a vyrobeného
zdravotnického prostředku, včetně kontroly
těch zdravotnických prostředků, které
požadované jakosti nedosáhnou (nebylo
u nich dosaženo shody),
3.2.2.3. pokud návrh, výrobu nebo výstupní kontrolu a zkoušky zdravotnických prostředků
nebo jejich částí provádí třetí osoba, metody sledování účinného fungování
systému jakosti a způsob a rozsah kontroly vykonávané nad třetí osobou,
3.2.3. postupů pro monitorování a ověřování návrhu zdravotnických prostředků, včetně
odpovídající
dokumentace, a to zejména
3.2.3.1. všeobecný popis zdravotnického prostředku, včetně všech plánovaných variant,
a jeho určená použití,
3.2.3.2. specifikace návrhu včetně norem, které se použijí, výsledky analýzy rizik
a popis
řešení přijatých pro splnění základních požadavků platných pro dané zdravotnické
prostředky, pokud se normy uvedené v § 4 odst. 2 neuplatňují v plném rozsahu,
3.2.3.3. techniky používané pro kontrolu a ověřování návrhu a postupy a systematická
opatření, která budou použita při navrhování zdravotnických prostředků,
3.2.3.4. pokud má být zdravotnický prostředek připojen k jinému zdravotnickému prostředku
nebo zdravotnickým prostředkům za účelem zamýšleného fungování, musí
být prokázáno, že splňuje základní požadavky, když je připojen k takovému zdravotnickému
prostředku nebo zdravotnickým prostředkům, které mají vlastnosti
uvedené výrobcem,
3.2.3.5. prohlášení, zda zdravotnický prostředek obsahuje jako svou integrální součást
látku nebo derivát z lidské krve uvedené v bodě 7.4. přílohy č. 1 k tomuto nařízení,
a údaje o zkouškách provedených v této souvislosti požadovaných k posouzení
bezpečnosti, jakosti a užitečnosti této látky nebo derivátu z lidské krve s přihlédnutím
k určenému účelu zdravotnického prostředku,
3.2.3.6. prohlášení, zda je zdravotnický prostředek vyroben s použitím tkání zvířecího
původu ve smyslu § 1 písm. b),
3.2.3.7. přijatá řešení, jak je uvedeno v části I. bodě 2. přílohy č. 1 k tomuto
nařízení,
3.2.3.8. preklinické hodnocení,
3.2.3.9. klinické hodnocení podle přílohy č. 10 k tomuto nařízení,
3.2.3.10. návrh značení a případně návod k použití,
3.2.4. techniky kontrol a zabezpečení jakosti ve stadiu
výroby zdravotnického prostředku, zejména
3.2.4.1. postupy a procedury, které budou použity
především pro sterilizaci a prodej a další
příslušné dokumenty,
3.2.4.2. postupy k identifikaci zdravotnického
prostředku vypracované a aktualizované ve
všech stadiích výroby na základě výkresů,
specifikací a dalších odpovídajících
dokumentů,
3.2.5. příslušné testy a zkoušky, které budou vykonány
před výrobou, během výroby a po výrobě
zdravotnických prostředků, jejich četnost a použitá
zkušební zařízení; zpětně musí být možné přiměřeným
způsobem zjistit správnost kalibrace zkušebních
zařízení.
3.3. Notifikovaná osoba provádí audity systému jakosti za
účelem zjištění, zda systém jakosti vyhovuje požadavkům
uvedeným v bodu 3.2. této přílohy. Shoda s těmito
požadavky se předpokládá u systémů jakosti, které
používají odpovídající harmonizované normy. V týmu pověřeném hodnocením systému
jakosti musí být alespoň 1 člen, který má zkušenost
s posuzováním technologie daného zdravotnického
prostředku. Součástí posouzení je posouzení
dokumentace návrhu příslušného zdravotnického prostředku
na reprezentativním základě, inspekční prohlídka
výrobních postupů v provozních prostorách výrobce
a v náležitě odůvodněných případech i kontrola
výrobních postupů v provozních prostorách dodavatelů
nebo dalších smluvních partnerů výrobce.
Notifikovaná osoba oznámí výrobci po provedeném auditu
systému jakosti jeho výsledek, který musí obsahovat závěry
kontroly a odůvodněné zhodnocení.
3.4. Výrobce informuje notifikovanou osobu, která schválila
systém jakosti o záměru, který podstatně mění systém
jakosti nebo jím pokrytý okruh zdravotnických prostředků.
Notifikovaná osoba zhodnotí navrhované změny, ověří, zda
takto změněný systém jakosti ještě vyhovuje požadavkům
uvedeným v bodu 3.2. této přílohy a své rozhodnutí, které
musí obsahovat závěry kontroly a odůvodněné zhodnocení,
oznámí výrobci.
4.
Přezkoumání návrhu zdravotnického prostředku.
4.1. Výrobce požádá, vedle svých úkolů podle bodu 3 této
přílohy, notifikovanou osobu o přezkoumání dokumentace
vztahující se k návrhu zdravotnického prostředku, který má
výrobce v úmyslu vyrábět a který patří do kategorie
zdravotnických prostředků podle bodu 3.1. této přílohy.
4.2. Žádost musí popisovat návrh a výrobu zdravotnického
prostředku, který má výrobce v úmyslu vyrábět, včetně
údajů o jeho účinnosti. K žádosti připojuje dokumenty,
které umožní posoudit, zda zdravotnický prostředek splňuje
požadavky podle bodu 3.2.3. této přílohy.
4.3. Působnost notifikované osoby
4.3.1. Notifikovaná osoba přezkoumá žádost, a jestliže zdravotnický prostředek vyhovuje
ustanovením tohoto nařízení, vydá žadateli certifikát ES přezkoumání návrhu. Notifikovaná
osoba může požádat, podle svého uvážení, o doplnění žádosti dalšími zkouškami nebo
důkazy, které jí umožní posoudit shodu zdravotnického prostředku s požadavky tohoto
nařízení. Certifikát ES přezkoumání návrhu musí obsahovat závěry přezkoumání, podmínky
platnosti certifikátu, údaje potřebné k identifikaci schváleného návrhu zdravotnického
prostředku, popřípadě popis určeného účelu použití.
4.3.2. V případě zdravotnických prostředků uvedených v bodě 7.4.1. přílohy č. 1 k
tomuto
nařízení požádá notifikovaná osoba, v souladu s bodem 7.4.2. přílohy č. 1 k tomuto
nařízení, před přijetím rozhodnutí o odborné stanovisko jeden z příslušných úřadů
členských
států, v České republice Ústav, nebo EMEA. Je-li o odborné stanovisko požádán
Ústav, vypracuje odborné stanovisko do 210 dnů ode dne obdržení úplné dokumentace.
Odborné stanovisko příslušného úřadu členského státu nebo EMEA musí být zahrnuto
do dokumentace týkající se zdravotnického prostředku. Notifikovaná osoba při přijímání
rozhodnutí věnuje tomuto odbornému stanovisku náležitou pozornost a své konečné
rozhodnutí sdělí instituci, která toto odborné stanovisko vydala.
4.3.3. V případě zdravotnických prostředků uvedených v bodě 7.4.3. přílohy č. 1 k
tomuto nařízení
musí být odborné stanovisko EMEA zahrnuto do dokumentace týkající se zdravotnického
prostředku. Notifikovaná osoba při přijímání rozhodnutí věnuje odbornému stanovisku
EMEA náležitou pozornost. Notifikovaná osoba nesmí vydat certifikát, pokud je
odborné stanovisko EMEA nepříznivé. Své konečné rozhodnutí sdělí EMEA.
4.3.4. V případě zdravotnických prostředků podle § 1 písm. b) vyráběných za použití
tkání zvířecího
původu musí notifikovaná osoba dodržovat postupy podle § 12.
4.4. Změny schváleného návrhu, které by mohly ovlivnit shodu se
základními požadavky tohoto nařízení nebo podmínky
předepsané pro použití zdravotnického prostředku,
podléhají dodatečnému schválení notifikovanou osobou,
která vydala certifikát ES přezkoumání návrhu. Vzhledem
k uvedenému žadatel informuje notifikovanou osobu, která
vydala certifikát ES přezkoumání návrhu, o všech změnách
schváleného návrhu.
Uvedené dodatečné schválení návrhu vydává notifikovaná
osoba jako dodatek k certifikátu ES přezkoumání návrhu.
5.
Dozor.
5.1. Cílem dozoru je zajistit, aby výrobce náležitě plnil
závazky vyplývající ze schváleného systému jakosti.
5.2. Výrobce zmocňuje notifikovanou osobu k provádění
nezbytných kontrol a poskytuje jí příslušné informace,
zejména
5.2.1. dokumentaci systému jakosti,
5.2.2. údaje požadované v části systému jakosti týkající se návrhu, jako jsou výsledky
analýz,
výpočtů, zkoušek, přijatá řešení, jak je uvedeno v části I. bodě 2. přílohy č. 1
k tomuto
nařízení, preklinické a klinické hodnocení, plán následného klinického sledování
po uvedení
na trh a popřípadě výsledky následného klinického sledování po uvedení na trh,
5.2.3. údaje vyhrazené systémem jakosti pro oblast výroby
zdravotnického prostředku, především zprávy
o kontrolách, zkouškách, kalibraci a o kvalifikaci
příslušných zaměstnanců.
5.3. Notifikovaná osoba
5.3.1. provádí periodicky příslušné kontroly a hodnocení
tak, aby se ujistila, že výrobce používá schválený
systém jakosti a poskytuje výrobci hodnotící
zprávu,
5.3.2. provádí, podle svého uvážení, u výrobce i předem
neohlášené kontroly, při nichž je oprávněna
v případě nezbytnosti provést nebo si vyžádat
provedení zkoušky za účelem kontroly, zda systém
jakosti je řádně činný.
O provedené kontrole, popřípadě i o výsledku
zkoušky, jestliže byla provedena, poskytuje výrobci
zprávu.
6.
Administrativní opatření.
6.1. Výrobce zdravotnického prostředku nebo jeho zplnomocněný zástupce musí po dobu
nejméně
5 let, a v případě implantabilních zdravotnických prostředků nejméně po dobu 15 let,
ode dne
výroby posledního zdravotnického prostředku uchovávat pro potřebu příslušných orgánů
státní
správy
6.1.1. písemné prohlášení o shodě,
6.1.2. dokumentaci systému jakosti podle bodu 3.1.4. této přílohy, a zejména dokumentaci,
údaje
a záznamy vzniklé na základě postupů uvedených v bodě 3.2.3.,
6.1.3. dokumentaci změn podle bodu 3.4.,
6.1.4. dokumentaci podle bodu 4.2.,
6.1.5. certifikáty a jiné dokumenty notifikované osoby podle bodů 3.3., 3.4., 4.3.,
4.4. a 5.3.
7.
Použití pro zdravotnické prostředky tříd IIa a IIb
7.1. V souladu s § 9 odst. 2 a 3 se tato příloha může používat na zdravotnické prostředky
tříd IIa a IIb.
Bod 4. se však nepoužije.
7.2. Pokud jde o zdravotnické prostředky třídy IIa, posoudí notifikovaná osoba jako
součást hodnocení
podle bodu 3.3. technickou dokumentaci nejméně 1 reprezentativního vzorku z každé
podskupiny
zdravotnických prostředků, jak je popsáno v bodě 3.2.3., s cílem posoudit soulad
s ustanoveními
tohoto nařízení.
7.3. Pokud jde o zdravotnické prostředky třídy IIb, posoudí notifikovaná osoba jako
součást hodnocení
podle bodu 3.3. technickou dokumentaci nejméně 1 reprezentativního vzorku z každé
skupiny
generických zdravotnických prostředků, jak je popsáno v bodě 3.2.3., s cílem posoudit
soulad
s ustanoveními tohoto nařízení.
7.4. Při volbě reprezentativního vzorku nebo vzorků bere notifikovaná osoba v úvahu
novost technologie,
podobnost návrhu, technologii, způsoby výroby a sterilizace, určené použití a výsledky
všech předchozích souvisejících posouzení (např. s ohledem na fyzikální, chemické
nebo biologické
vlastnosti). Notifikovaná osoba zdokumentuje a uchovává pro potřebu příslušných orgánů
státní
správy důvody volby vzorku nebo vzorků.
7.5. Další vzorky posuzuje notifikovaná osoba jako součást dozoru podle bodu 5.
8.
Po dokončení výroby každé šarže zdravotnických prostředků
uvedených v § 2 odst. 2 písm. g) zákona o zdravotnických
prostředcích uvědomí výrobce notifikovanou osobu o uvolnění
šarže těchto prostředků a zašle jí certifikát týkající se
uvolnění šarže derivátu z lidské krve použitého v tomto
prostředku, vydaný příslušnou laboratoří určenou k tomuto účelu
členským státem, v České republice
v souladu se zvláštním právním předpisem.26)
Příl.3
ES PŘEZKOUŠENÍ TYPU
1.
ES přezkoušení typu zdravotnického prostředku je postup, kterým
notifikovaná osoba zjišťuje a certifikuje, že reprezentativní
vzorek posuzované výroby zdravotnických prostředků (dále jen
"typ") splňuje příslušná ustanovení tohoto nařízení, která se
na něj vztahují. Ve spojitosti s tím výrobce zaručuje
a prohlašuje, že zdravotnické prostředky dodávané na trh
odpovídají certifikovanému typu.
2.
Výrobce, popřípadě zplnomocněný zástupce požádá notifikovanou
osobu o přezkoušení typu zdravotnického prostředku; žádost
obsahuje:
2.1. jméno, popřípadě jména a příjmení výrobce, adresu jeho
bydliště a místa podnikání nebo adresu provozovny,
jestliže výrobcem je fyzická osoba žijící v zahraničí,
jméno, popřípadě jména a příjmení, adresu trvalého pobytu
výrobce, jestliže má trvalý pobyt v České republice;
obchodní firmu nebo název, sídlo a adresu provozovny,
jde-li o právnickou osobu,
2.2. jméno, popřípadě jména a příjmení a adresu bydliště
zplnomocněného zástupce, jde-li o fyzickou osobu; obchodní
firmu nebo název a adresu sídla, jestliže podává žádost
právnická osoba,
2.3. dokumentaci podle bodu 3 této přílohy, která je potřebná
k posouzení shody typu s požadavky tohoto nařízení.
Žadatel předá typ notifikované osobě, která si podle svého
uvážení vyžádá další vzorky,
2.4. písemné prohlášení, že stejná žádost nebyla podána jiné
notifikované osobě.
3.
Dokumentace musí umožňovat pochopení návrhu, výroby a funkční způsobilosti zdravotnického
prostředku
a musí obsahovat zejména:
3.1. celkový popis typu, včetně všech plánovaných variant, a jeho určená použití,
3.2. konstrukční výkresy, předpokládané výrobní technologie, zejména pokud jde o
sterilizaci, a schémata
součástí, podsestav, obvodů atd.,
3.3. popisy a vysvětlivky potřebné pro pochopení výše uvedených výkresů schémat a
fungování zdravotnického
prostředku,
3.4. seznam norem uvedených v § 4 odst. 2 používaných plně nebo částečně a popis
řešení přijatých pro
splnění základních požadavků, pokud se normy uvedené v § 4 odst. 2 neuplatňují v
plném rozsahu,
3.5. výsledky provedených konstrukčních výpočtů, analýzy rizik, výzkumů, technických
zkoušek atd.,
3.6. prohlášení, zda zdravotnický prostředek obsahuje jako svou integrální součást
látku nebo derivát
z lidské krve uvedené v bodě 7.4. přílohy č. 1 k tomuto nařízení, a údaje o zkouškách
provedených
v této souvislosti požadovaných k posouzení bezpečnosti, jakosti a užitečnosti této
látky nebo
derivátu z lidské krve s přihlédnutím k určenému účelu použití zdravotnického prostředku,
3.7. prohlášení, zda je zdravotnický prostředek vyroben s použitím tkání zvířecího
původu, ve smyslu
§ 1 písm. b),
3.8. přijatá řešení, jak je uvedeno v části I. bodě 2. přílohy č. 1 k tomuto nařízení,
3.9. preklinické hodnocení,
3.10. klinické hodnocení podle přílohy č. 10 k tomuto nařízení,
3.11. návrh značení a případně návod k použití.
4.
Notifikovaná osoba
4.1. přezkoumá a zhodnotí dokumentaci a ověří, zda byl typ
vyroben v souladu s touto dokumentací. O požadavcích,
které jsou navrhovány v souladu s příslušnými
harmonizovanými normami uvedenými v § 4 odst. 2,
a o skutečnostech, které nejsou navrhovány podle těchto
norem, pořídí záznam,
4.2. provede nebo nechá provést příslušné kontroly a zkoušky
nezbytné k ověření, zda řešení přijatá výrobcem splňují
požadavky uvedené v § 4 odst. 1 tohoto nařízení, a to
v případě nepoužití norem uvedených v § 4 odst. 2.
Jestliže pro určený účel použití má být zdravotnický
prostředek spojen s jiným zdravotnickým prostředkem,
popřípadě jinými zdravotnickými prostředky, musí být
prokázáno, že systém nebo souprava jako celek vyhovuje
základním požadavkům, jestliže je připojen k takovému
prostředku nebo prostředkům, pokud má (mají)
charakteristiky specifikované výrobcem,
4.3. provede nebo nechá provést příslušné kontroly nebo zkoušky
nezbytné k ověření, zda výrobce skutečně použil
odpovídající harmonizované normy, které zvolil,
4.4. dohodne se žadatelem místo, kde budou nezbytné kontroly
a zkoušky prováděny,
5.
Podmínky vydání dokumentů notifikovanou osobou
5.1. Jestliže typ splňuje ustanovení tohoto nařízení, vydá notifikovaná osoba žadateli
certifikát ES přezkoušení
typu. K certifikátu ES přezkoušení typu musí být přiloženy odpovídající části dokumentace,
jejichž 1 kopii uchovává notifikovaná osoba. Certifikát ES přezkoušení typu musí
obsahovat:
5.1.1. jméno, popřípadě jména, a příjmení výrobce, adresu jeho bydliště a místa podnikání,
jestliže
výrobcem je fyzická osoba žijící v zahraničí, jméno, popřípadě jména, a příjmení,
adresu
trvalého pobytu výrobce, jestliže má trvalý pobyt v České republice; obchodní firmu
nebo
název, sídlo a adresu provozovny, jde-li o právnickou osobu,
5.1.2. závěry posouzení, podmínky platnosti a údaje potřebné k identifikaci schváleného
typu.
5.2. V případě zdravotnických prostředků uvedených v bodě 7.4.1. přílohy č. 1 k tomuto
nařízení
požádá notifikovaná osoba, v souladu s bodem 7.4.2. přílohy č. 1 k tomuto nařízení,
před přijetím
rozhodnutí o odborné stanovisko jeden z příslušných úřadů členských států, v České
republice
Ústav, nebo EMEA. Je-li o odborné stanovisko požádán Ústav, vypracuje odborné stanovisko
do
210 dnů ode dne obdržení úplné dokumentace. Odborné stanovisko příslušného úřadu
členského
státu nebo EMEA musí být zahrnuto do dokumentace týkající se zdravotnického prostředku.
Notifikovaná osoba při přijímání rozhodnutí věnuje tomuto odbornému stanovisku náležitou
pozornost a své konečné rozhodnutí sdělí instituci, která toto odborné stanovisko
vydala.
5.3. V případě zdravotnických prostředků uvedených v bodě 7.4.3. přílohy č. 1 k tomuto
nařízení musí
být odborné stanovisko EMEA zahrnuto do dokumentace týkající se zdravotnického prostředku.
Notifikovaná osoba při přijímání rozhodnutí věnuje odbornému stanovisku EMEA náležitou
pozornost.
Notifikovaná osoba nesmí vydat certifikát, pokud je odborné stanovisko EMEA nepříznivé.
Své konečné rozhodnutí sdělí EMEA.
5.4. V případě zdravotnických prostředků podle § 1 písm. b) vyráběných za použití
tkání zvířecího
původu notifikovaná osoba dodržuje postupy podle § 12.
6.
Výrobce, popřípadě zplnomocněný zástupce informuje notifikovanou osobu, která
vydala certifikát ES
přezkoušení typu o jakékoliv významné změně provedené na schváleném zdravotnickém
prostředku.
Změny schváleného zdravotnického prostředku musí získat dodatečný souhlas od notifikované
osoby,
která vydala certifikát ES přezkoušení typu, pokud změny mohou ovlivnit shodu se
základními požadavky
nebo s podmínkami stanovenými pro použití zdravotnického prostředku. V takovém případě
má
dodatečný souhlas formu dodatku k původnímu certifikátu ES přezkoušení typu.
7.
Administrativní opatření
7.1. Notifikované osoby mohou obdržet kopie certifikátů ES přezkoušení typu, popřípadě
jejich dodatků.
Přílohy těchto certifikátů musí být dostupné jiným notifikovaným osobám na základě
odůvodněné
žádosti po předchozím informování výrobce.
7.2. Výrobce nebo jeho zplnomocněný zástupce uchovává spolu s technickou dokumentací
kopie certifikátů
ES přezkoušení typu a jejich dodatků po dobu nejméně 5 let ode dne výroby posledního
zdravotnického prostředku. V případě implantabilních zdravotnických prostředků činí
tato doba
nejméně 15 let ode dne výroby posledního zdravotnického prostředku.
Příl.4
ES OVĚŘOVÁNÍ
1.
ES ověřování je postup, kterým výrobce nebo jeho zplnomocněný
zástupce zajišťuje a prohlašuje, že zdravotnické prostředky,
které byly přezkoušeny notifikovanou osobou podle bodu 4 této
přílohy, odpovídají typu popsanému v certifikátu ES přezkoušení
typu a vyhovují ustanovením, která se na ně vztahují z tohoto
nařízení.
2.
Postup výrobce.
Výrobce
2.1. provede nezbytná opatření, aby zajistil, že vyráběné
zdravotnické prostředky odpovídají typu uvedenému
v certifikátu ES přezkoušení typu a vyhovují požadavkům,
které se na ně vztahují z tohoto nařízení,
2.2. připraví, před zahájením výroby, dokumenty vymezující
2.2.1. výrobní proces, zejména pokud jde o sterilizaci,
jestliže je to nutné,
2.2.2. rutinní postupy s předběžnými opatřeními, která
mají být zavedena k zajištění stejnorodosti výroby,
2.2.3. podle potřeby i soulad zdravotnických prostředků
s typem popsaným v certifikátu ES přezkoušení typu
a s požadavky, které se na ně vztahují z tohoto
nařízení,
2.3. připojí označení CE v souladu s § 5 a vypracuje písemné
prohlášení o shodě,
2.4. zajišťuje a udržuje sterilitu při výrobě zdravotnických
prostředků uváděných na trh ve sterilním stavu, za tím
účelem postupuje kromě výše uvedeného i podle bodů 3 a 4
přílohy č. 5 k tomuto nařízení.
3.
Výrobce zavede a bude aktualizovat systematický postup vyhodnocování zkušeností
získaných s vyrobenými
zdravotnickými prostředky, včetně ustanovení uvedených v příloze č. 10 k tomuto nařízení,
a vhodným způsobem zavádět nezbytná nápravná opatření; součástí tohoto závazku je
povinnost výrobce
oznámit Ústavu nežádoucí příhody30), jakmile se o nich dozví.
4.
Notifikovaná osoba
4.1. provede odpovídající přezkoumání a zkoušky k ověření shody
zdravotnického prostředku s požadavky tohoto nařízení
podle rozhodnutí výrobce u
4.1.1. každého zdravotnického prostředku podle bodu 5 této
přílohy nebo
4.1.2. zdravotnických prostředků vybraných statisticky
podle bodu 6 této přílohy.
Uvedené kontroly neplatí pro hlediska výrobního postupu,
která jsou určena pro zajištění sterility.
5.
Ověřování přezkoumáním a zkouškami každého zdravotnického
prostředku.
Notifikovaná osoba
5.1. zkouší každý zdravotnický prostředek individuálně, za
účelem ověření shody každého zdravotnického prostředku
s typem uvedeným v certifikátu ES přezkoušení typu
a s požadavky, které se na ně vztahují z tohoto nařízení.
Notifikovaná osoba provádí odpovídající zkoušky stanovené
v příslušné harmonizované normě (normách) a v případě
potřeby provádí ekvivalentní zkoušky,
5.2. umístí nebo nechá umístit na každý schválený zdravotnický
prostředek své identifikační číslo a s odvoláním na
provedené zkoušky vypracuje písemný certifikát o shodě
každého zdravotnického prostředku s typem uvedeným
v certifikátu ES přezkoušení typu a s požadavky
vyplývajícími pro ně z tohoto nařízení.
6.
Statistické ověřování.
6.1. Výrobce předloží zdravotnické prostředky vyrobené ve
stejnorodých výrobních dávkách (šaržích).
6.2. Notifikovaná osoba odebere z každé výrobní dávky (šarže)
náhodně vybraný vzorek, jednotlivé zdravotnické prostředky
z tohoto vzorku zkoumá individuálně, přičemž postupuje
podle bodu 5 této přílohy, popřípadě se jednotlivé
zdravotnické prostředky podrobí ekvivalentním zkouškám
k ověření shody těchto zdravotnických prostředků s typem
popsaným v certifikátu ES přezkoušení typu a s požadavky
tohoto nařízení, které se na ně vztahují, za účelem
stanovení, zda má být výrobní dávka (šarže) schválena,
nebo neschválena.
6.3. Statistické ověřování zdravotnických prostředků srovnáváním nebo měřením využívá
přejímací
plány s operativními charakteristikami, které zajišťují vysokou úroveň bezpečnosti
a funkční způsobilosti
podle nejnovějších poznatků vědy a techniky. Přejímací plány jsou stanoveny podle
harmonizovaných norem podle § 4 odst. 2 s přihlédnutím k povaze daných skupin zdravotnických
prostředků.
6.4. Jestliže notifikovaná osoba výrobní dávku (šarži) schválí,
umístí nebo nechá umístit na každý zdravotnický prostředek
své identifikační číslo a vypracuje písemný certifikát
o shodě s odvoláním na provedené zkoušky. Všechny
zdravotnické prostředky z výrobní dávky (šarže) mohou být
uvedeny na trh s výjimkou zdravotnických prostředků ve
vybraném vzorku, které byly shledány nevyhovujícími.
Jestliže notifikovaná osoba výrobní dávku (šarži)
neschválí, učiní opatření, aby zabránila uvedení výrobní
dávky (šarže) na trh.
Notifikovaná osoba může pozastavit statistické ověřování,
v případě častého neschválení výrobních dávek (šarží).
Výrobce může umisťovat na zdravotnický prostředek
identifikační číslo notifikované osoby na její odpovědnost
již v průběhu výrobního procesu.
7.
Administrativní opatření.
Výrobce zdravotnického
prostředku nebo jeho zplnomocněný zástupce musí
po dobu nejméně 5 let, a v případě implantabilních
zdravotnických prostředků nejméně po dobu 15 let,
ode dne výroby posledního zdravotnického prostředku
uchovávat pro potřebu příslušných orgánů státní
správy
7.1. prohlášení o shodě,
7.2. dokumentaci uvedenou v bodu 2 této přílohy,
7.3. certifikáty uvedené v bodech 5.2. a 6.4. této přílohy,
popřípadě
7.4. certifikát ES přezkoušení typu podle přílohy č. 3
k tomuto nařízení.
8.
Použití u zdravotnických prostředků třídy IIa.
V souladu s § 9 odst. 2 lze tuto přílohu použít pro zdravotnické
prostředky třídy IIa s tím, že odchylně od
8.1. bodů 1 a 2 této přílohy na základě prohlášení o shodě
výrobce zajišťuje a prohlašuje, že zdravotnické prostředky
třídy IIa jsou vyrobeny v souladu s technickou dokumentací
podle bodu 3 přílohy č. 7 k tomuto nařízení a vyhovují
požadavkům, které se na ně vztahují z tohoto nařízení,
8.2. bodů 1, 2, 5 a 6 této přílohy jsou ověření provedená
notifikovanou osobou určena k potvrzení shody
zdravotnických prostředků třídy IIa s technickou
dokumentací podle bodu 3 přílohy č. 7 k tomuto nařízení.
9.
Použití pro zdravotnické prostředky uvedené v § 2 odst. 2 písm. g)
zákona o zdravotnických prostředcích.
V případě ověřování podle bodu 5 této přílohy, po dokončení
výroby každé šarže zdravotnických prostředků uvedených v
§ 2 odst. 2 písm. g) zákona o zdravotnických prostředcích,
a v případě ověřování podle bodu 6 této přílohy výrobce uvědomí
notifikovanou osobu o uvolnění této šarže zdravotnických
prostředků a zašle jí úřední certifikát týkající se uvolnění
šarže derivátu z lidské krve použitého ve zdravotnickém
prostředku vydaný příslušnou laboratoří určenou k tomuto účelu
členským státem, v České republice
v souladu se zvláštním právním předpisem.26)
Příl.5
ES PROHLÁŠENÍ O SHODĚ
(Zabezpečení jakosti výroby)
1.
Výrobce zajišťuje zavedení a provozování systému jakosti pro výrobu a výstupní
kontrolu příslušných
zdravotnických prostředků podle bodu 3., systém jakosti podléhá auditu podle bodu
3. a podléhá dozoru
podle bodu 4.
2.
ES prohlášení o shodě (Zabezpečení jakosti výroby) je součástí postupu, jímž výrobce,
který plní povinnosti
podle bodu 1., zajišťuje a prohlašuje, že příslušné zdravotnické prostředky odpovídají
typu
popsanému v certifikátu ES přezkoušení typu a vyhovují ustanovením tohoto nařízení,
která se na ně
vztahují.
Výrobce
2.1. opatřuje zdravotnické prostředky označením CE v souladu s § 5 a
2.2. vypracuje písemné prohlášení o shodě, které je povinen uchovávat; toto prohlášení
se vypracuje pro
1 nebo více vyrobených zdravotnických prostředků jasně identifikovaných názvem zdravotnického
prostředku, kódem zdravotnického prostředku nebo jiným jednoznačným odkazem.
3.
Systém jakosti.
3.1. Výrobce
3.1.1. předkládá písemnou žádost notifikované osobě
o posouzení (vyhodnocení a schválení) svého systému
jakosti; žádost musí obsahovat
3.1.1.1. jméno, popřípadě jména a příjmení výrobce,
adresu jeho bydliště a místa podnikání,
jestliže výrobcem je fyzická osoba žijící
v zahraničí, jméno, popřípadě jména
a příjmení, adresu trvalého pobytu
výrobce, jestliže má trvalý pobyt v České
republice; obchodní firmu nebo název,
sídlo a adresu provozovny, jde-li
o právnickou osobu,
3.1.1.2. významné informace o zdravotnickém
prostředku nebo kategorii zdravotnických
prostředků, pro které platí postup uvedený
v bodu 1,
3.1.1.3. písemné prohlášení, že u jiné notifikované
osoby nebyla podána žádost o posouzení
systému jakosti výroby vztahující se ke
stejnému zdravotnickému prostředku,
3.1.1.4. dokumentaci systému jakosti,
3.1.1.5. záruku výrobce plnit požadavky vyplývající
ze schváleného systému jakosti,
3.1.1.6. záruku výrobce udržovat schválený systém
jakosti v použitelném a účinném stavu,
3.1.1.7. v případě potřeby technickou dokumentaci
schválených typů zdravotnických prostředků
a kopii certifikátu ES přezkoušení typu,
3.1.1.8. závazek výrobce, že zavede a bude aktualizovat systematický postup vyhodnocování
zkušeností získaných s vyrobenými zdravotnickými prostředky, včetně ustanovení
uvedených v příloze č. 10 k tomuto nařízení, a vhodným způsobem zavádět
nezbytná nápravná opatření; součástí tohoto závazku je povinnost výrobce oznámit
Ústavu nežádoucí příhody podle zákona o zdravotnických prostředcích30) ihned,
jakmile se o nich dozví.
3.2. Uplatnění systému jakosti musí zajistit, že vyrobené
zdravotnické prostředky odpovídají typu popsanému
v certifikátu ES přezkoušení typu.
Prvky, požadavky a ustanovení schválené výrobcem pro
systém jakosti musí být systematicky a řádně dokumentovány
v písemně vypracovaných zásadách a postupech. Tato
dokumentace systému jakosti musí umožnit jednotný výklad
koncepce jakosti zdravotnických prostředků a postupů,
zejména programů jakosti, plánů jakosti, příruček jakosti
a záznamů o jakosti.
Dokumentace systému jakosti musí obsahovat zejména popis
3.2.1. cílů jakosti u výrobce,
3.2.2. organizace výrobce, především
3.2.2.1. organizačních struktur, zodpovědnosti
vedoucích zaměstnanců a jejich pravomocí
ve vztahu k výrobě zdravotnických
prostředků,
3.2.2.2. metod sledování účinnosti systému jakosti,
zejména jeho schopnosti dosáhnout
požadované jakosti zdravotnického
prostředku, včetně kontroly zdravotnických
prostředků, které požadované jakosti
nedosáhnou,
3.2.2.3. pokud výrobu nebo výstupní kontrolu a zkoušky zdravotnických prostředků
nebo
jejich částí provádí třetí osoba, metody sledování účinného fungování systému
jakosti, a zejména způsob a rozsah kontroly vykonávané nad třetí osobou,
3.2.3. techniky kontrol a zajištění jakosti zdravotnických
prostředků ve stadiu jejich výroby, zejména
3.2.3.1. postupy, které budou použity především
pokud jde o sterilizaci, prodej
a příslušné dokumenty,
3.2.3.2. postupy k identifikaci zdravotnického
prostředku vypracované a aktualizované ke
všem stadiím výroby na základě výkresů,
specifikací a dalších odpovídajících
dokumentů,
3.2.4. příslušné testy a zkoušky, které budou vykonány
před výrobou, během výroby a po výrobě
zdravotnických prostředků, jejich četnost a použitá
zkušební zařízení; zpětně musí být možné přiměřeným
způsobem zjistit správnost kalibrace zkušebních
zařízení.
3.3. Notifikovaná osoba provádí audity systému jakosti za
účelem zjištění, zda systém jakosti vyhovuje požadavkům
uvedeným v bodu 3.2. této přílohy. Shoda s těmito
požadavky se předpokládá u systémů jakosti, u kterých se
používají odpovídající harmonizované normy. V týmu
pověřeném hodnocením systému jakosti musí být alespoň
jeden člen, který má zkušenosti s hodnocením příslušných
technologií. Hodnotitelský postup zahrnuje prohlídku
provozních prostor výrobce a v odůvodněných případech
i kontrolu výrobních postupů v provozních prostorech
dodavatelů výrobce, popřípadě jeho dalších smluvních
stran.
Notifikovaná osoba oznámí výrobci, po provedeném auditu
systému jakosti, jeho výsledek, který musí obsahovat
závěry kontroly a odůvodněné zhodnocení.
3.4. Výrobce informuje notifikovanou osobu, která schválila
systém jakosti, o záměru, který podstatně mění systém
jakosti nebo jím pokrytý okruh zdravotnických prostředků.
Notifikovaná osoba zhodnotí navrhované změny, ověří, zda
takto změněný systém jakosti ještě vyhovuje požadavkům
uvedeným v bodu 3.2. této přílohy a své rozhodnutí, které
musí obsahovat závěry kontroly a odůvodněné zhodnocení,
oznámí výrobci.
4.
Dozor.
4.1. Cílem dozoru je zajistit, aby výrobce náležitě plnil
požadavky vyplývající ze schváleného systému jakosti.
4.2. Za účelem uvedeným v bodu 4.1. této přílohy výrobce
zmocňuje notifikovanou osobu k provádění nezbytných
kontrol a poskytuje jí příslušné informace, zejména
4.2.1. dokumentaci systému jakosti,
4.2.2. technickou dokumentaci,
4.2.3. údaje vymezené systémem jakosti pro oblast výroby
zdravotnických prostředků, především kontrolní
zprávy, údaje o zkouškách, kalibraci a zprávy
o kvalifikaci příslušných zaměstnanců.
4.3. Notifikovaná osoba provádí
4.3.1. periodicky příslušné kontroly a hodnocení tak, aby
se ujistila, že výrobce používá schválený systém
jakosti; výrobci poskytuje o provedené kontrole
hodnotící zprávu,
4.3.2. podle svého uvážení u výrobce i předem neohlášené
kontroly, při nichž v případě potřeby provede nebo
nechá provést zkoušky za účelem kontroly, zda
systém jakosti je řádně činný; o provedené
kontrole, a o výsledku zkoušky, jestliže byla
provedena, poskytuje výrobci písemnou zprávu.
5.
Administrativní opatření.
5.1. Výrobce zdravotnického prostředku nebo jeho zplnomocněný zástupce musí po dobu
nejméně
5 let, a v případě implantabilních zdravotnických prostředků nejméně po dobu 15 let,
ode dne
výroby posledního zdravotnického prostředku uchovávat pro potřebu příslušných orgánů
státní
správy
5.1.1. prohlášení o shodě,
5.1.2. dokumentaci systému jakosti,
5.1.3. změny podle bodu 3.4. této přílohy,
5.1.4. technickou dokumentaci schválených typů a kopie
certifikátů ES přezkoušení typu,
5.1.5. rozhodnutí a zprávy notifikované osoby podle bodů
3.4. a 4.3. této přílohy,
5.1.6. certifikát ES přezkoušení typu podle přílohy č. 3 k tomuto nařízení.
6.
Použití pro zdravotnické prostředky třídy IIa
V souladu s § 9 odst. 2 lze tuto přílohu použít na zdravotnické prostředky třídy
IIa s těmito odchylkami:
6.1. Odchylně od bodů 2., 3.1. a 3.2. na základě prohlášení o shodě výrobce zajišťuje
a prohlašuje, že jsou
zdravotnické prostředky třídy IIa vyrobeny ve shodě s technickou dokumentací podle
bodu 3.
přílohy č. 7 k tomuto nařízení a splňují požadavky tohoto nařízení, které se na ně
vztahují.
6.2. Pokud jde o zdravotnické prostředky třídy IIa, posoudí notifikovaná osoba jako
součást hodnocení
podle bodu 3.3. technickou dokumentaci podle bodu 3. přílohy č. 7 k tomuto nařízení
nejméně
1 reprezentativního vzorku z každé podskupiny zdravotnických prostředků s cílem posoudit
soulad
s ustanoveními tohoto nařízení.
6.3. Při volbě reprezentativního vzorku nebo vzorků bere notifikovaná osoba v úvahu
novost technologie,
podobnost návrhu, technologii, způsoby výroby a sterilizace, určené použití a výsledky
všech předchozích souvisejících hodnocení (např. s ohledem na fyzikální, chemické
a biologické
vlastnosti), která byla provedena v souladu s tímto nařízením. Notifikovaná osoba
zdokumentuje
a uchovává pro potřebu příslušného orgánu státní správy důvody volby vzorku nebo
vzorků.
6.4. Další vzorky posuzuje notifikovaná osoba jako součást dozoru podle bodu 4.3.
7.
Po dokončení výroby každé šarže zdravotnických prostředků
uvedených v § 2 odst. 2 písm. g) zákona o zdravotnických
prostředcích uvědomí výrobce notifikovanou osobu o uvolnění
šarže těchto prostředků a zašle jí úřední certifikát týkající
se uvolnění šarže derivátu z lidské krve použitého ve
zdravotnickém prostředku, vydaný příslušnou laboratoří určenou
k tomuto účelu členským státem,
v České republice v souladu se zvláštním právním předpisem.26)
Příl.6
ES PROHLÁŠENÍ O SHODĚ
(Zabezpečení jakosti zdravotnického prostředku)
1.
Výrobce zajišťuje zavedení a provozování systému jakosti schváleného pro výstupní
kontrolu a zkoušky
příslušných zdravotnických prostředků podle bodu 3. a podléhá dozoru podle bodu 4.
V případě zdravotnických
prostředků uváděných na trh ve sterilním stavu uplatňuje ustanovení bodů 3. a 4.
přílohy č. 5
k tomuto nařízení, a to pouze pro vlastnosti výrobního procesu určené pro zajištění
a udržení sterility.
2.
ES prohlášení o shodě (Zabezpečení jakosti zdravotnického prostředku) je částí
postupu, jímž výrobce,
který plní povinnosti podle bodu 1., zajišťuje a prohlašuje, že příslušné zdravotnické
prostředky
odpovídají typu popsanému v certifikátu ES přezkoušení typu a vyhovují ustanovením
tohoto nařízení,
která se na ně vztahují.
Výrobce
2.1. opatřuje zdravotnické prostředky označením CE v souladu s § 5, kdy označení
CE je doplněno
identifikačním číslem notifikované osoby, která je zapojena do posuzování zdravotnických
prostředků
podle této přílohy, a
2.2. vypracuje písemné prohlášení o shodě, které je povinen uchovávat; toto prohlášení
se vypracuje pro
1 nebo více vyrobených zdravotnických prostředků jasně identifikovaných názvem zdravotnického
prostředku, kódem zdravotnického prostředku nebo jiným jednoznačným odkazem.
3.
Systém jakosti.
3.1. Výrobce
3.1.1. předkládá písemnou žádost notifikované osobě
o posouzení (vyhodnocení a schválení) svého systému
jakosti; žádost musí obsahovat
3.1.1.1. jméno, popřípadě jména a příjmení výrobce,
adresu jeho bydliště a místa podnikání,
jestliže výrobcem je fyzická osoba žijící
v zahraničí, jméno, popřípadě jména
a příjmení, adresu trvalého pobytu
výrobce, jestliže má trvalý pobyt v České
republice; obchodní firmu nebo název,
sídlo a adresu provozovny, jde-li
o právnickou osobu,
3.1.1.2. potřebné informace o zdravotnickém
prostředku nebo o kategorii zdravotnických
prostředků, pro které platí postup uvedený
v bodu 1,
3.1.1.3. písemné prohlášení, že u jiné notifikované
osoby nebyla podána žádost o posouzení
systému jakosti pro stejné zdravotnické
prostředky,
3.1.1.4. dokumentaci systému jakosti,
3.1.1.5. záruku výrobce plnit požadavky vyplývající
ze schváleného systému jakosti,
3.1.1.6. záruku výrobce udržovat schválený systém
jakosti na odpovídající a efektivní
úrovni,
3.1.1.7. v případě potřeby technickou dokumentaci
schválených typů a kopii certifikátu ES
přezkoušení typu,
3.1.1.8. závazek výrobce, že zavede a bude aktualizovat systematický postup vyhodnocování
zkušeností získaných s vyrobenými zdravotnickými prostředky, včetně ustanovení
uvedených v příloze č. 10 k tomuto nařízení, a vhodným způsobem zavádět
nezbytná nápravná opatření; součástí tohoto závazku je povinnost výrobce oznámit
Ústavu nežádoucí příhody podle zákona o zdravotnických prostředcích30) ihned,
jakmile se o nich dozví.
3.2. Výrobce v rámci tohoto systému jakosti
3.2.1. přezkoumá každý zdravotnický prostředek nebo
reprezentativní vzorek z každé výrobní dávky
(šarže),
3.2.2. provede příslušné zkoušky stanovené v odpovídající
harmonizované normě (normách), popřípadě
ekvivalentní zkoušky, aby se ujistil, že
zdravotnické prostředky odpovídají typu popsanému
v certifikátu ES přezkoušení typu a vyhovují
ustanovením, která se na ně vztahují z tohoto
nařízení,
3.2.3. systematicky a řádně dokumentuje prvky, požadavky
a ustanovení jím přijaté; formou písemných
opatření, postupů a pokynů tato dokumentace musí
umožnit jednotný výklad programu jakosti, plánu
jakosti, příruček jakosti a záznamů o jakosti.
Tato dokumentace musí zahrnovat zejména
odpovídající popis
3.2.3.1. cílů jakosti u výrobce,
3.2.3.2. organizačních struktur, odpovědnosti
a pravomoci vedoucích zaměstnanců
z hlediska jakosti výroby,
3.2.3.3. přezkoumání a zkoušek zdravotnických
prostředků po jejich vyrobení; zpětně musí
být možné přiměřeným způsobem zjistit
správnost kalibrace zkušebních zařízení,
3.2.3.4. metod sledování účinnosti systému jakosti,
3.2.3.5. záznamů o jakosti, především kontrolních
zpráv, zpráv o zkouškách a kalibraci
a o kvalifikaci příslušných zaměstnanců,
3.2.3.6. pokud výrobu nebo výstupní kontrolu a zkoušky zdravotnických prostředků
nebo
jejich částí provádí třetí osoba, metody sledování účinného fungování systému
jakosti, a způsob a rozsah kontroly vykonávané nad třetí osobou.
Uvedené kontroly neplatí pro oblasti výrobního
procesu určené k zajištění sterility.
3.3. Notifikovaná osoba
3.3.1. provádí audity systému jakosti u výrobce s cílem
zjistit, zda tento systém vyhovuje požadavkům podle
bodu 3.2. této přílohy, shodu s těmito požadavky
předpokládá u systémů jakosti, ve kterých se
používají harmonizované normy.
V týmu pověřeném hodnocením systému jakosti musí
být alespoň jeden člen, který má zkušenosti
s hodnocením příslušných technologií. Hodnotitelský
postup zahrnuje kontrolu provozních prostor výrobce
a v odůvodněných případech i kontrolu výrobních
postupů v provozních prostorech dodavatelů výrobce.
Notifikovaná osoba oznámí výrobci po provedeném
auditu systému jakosti jeho výsledek (rozhodnutí),
který musí obsahovat závěry kontroly a odůvodněné
zhodnocení.
3.4. Výrobce informuje notifikovanou osobu, která schválila
systém jakosti, o záměru podstatně změnit tento systém.
Notifikovaná osoba zhodnotí navrhované změny a ověří, zda
takto změněný systém jakosti ještě vyhovuje požadavkům
uvedeným v bodu 3.2. této přílohy a oznámí své rozhodnutí
výrobci; toto rozhodnutí musí obsahovat závěry kontroly
a odůvodněné zhodnocení.
4.
Dozor.
4.1. Cílem dozoru je zajistit, aby výrobce náležitě plnil
požadavky vyplývající ze schváleného systému jakosti.
4.2. Za účelem uvedeným v bodu 4.1. této přílohy výrobce
4.2.1. umožňuje notifikované osobě přístup do kontrolních,
zkušebních a skladovacích prostorů, které používá
v rámci výroby zdravotnických prostředků,
4.2.2. poskytuje notifikované osobě příslušné informace,
zejména
4.2.2.1. dokumentaci systému jakosti,
4.2.2.2. technickou dokumentaci,
4.2.2.3. záznamy o jakosti, především kontrolní
zprávy, údaje o zkouškách a kalibraci
a o kvalifikaci příslušných zaměstnanců.
4.3. Notifikovaná osoba
4.3.1. provádí periodicky příslušné kontroly a hodnocení
tak, aby se ujistila, že výrobce používá schválený
systém jakosti a
4.3.2. poskytuje výrobci hodnotící zprávu.
4.4. Notifikovaná osoba provádí u výrobce podle svého uvážení
i předem neohlášené kontroly, při nichž v případě potřeby
provede nebo si vyžádá provedení zkoušky za účelem
kontroly, zda systém jakosti je řádně činný a výroba
zdravotnických prostředků odpovídá požadavkům, které se na
ně vztahují z tohoto nařízení.
Při těchto kontrolách se ověřuje odpovídající vzorek
vyrobených zdravotnických prostředků odebraný u výrobce
notifikovanou osobou a provedou se příslušné zkoušky podle
odpovídajících harmonizovaných norem nebo se provádí
ekvivalentní zkoušky. Jestliže se neshoduje jeden nebo
více vzorků, učiní notifikovaná osoba příslušná opatření.
Notifikovaná osoba poskytne výrobci zprávu o kontrole
a jestliže byly provedeny zkoušky i zkušební zprávu.
5.
Administrativní opatření.
5.1. Výrobce zdravotnického prostředku nebo jeho zplnomocněný zástupce musí po dobu
nejméně
5 let, a v případě implantabilních zdravotnických prostředků nejméně po dobu 15 let,
ode dne
výroby posledního zdravotnického prostředku uchovávat pro potřebu příslušných orgánů
státní
správy
5.1.1. prohlášení o shodě,
5.1.2. dokumentaci systému jakosti podle bodu 3.1.1.4.,
5.1.3. změny podle bodu 3.4.,
5.1.4. rozhodnutí a zprávy notifikované osoby podle bodů 3.4., 4.3. a 4.4.,
5.1.5. certifikát ES přezkoušení typu podle přílohy č. 3 k tomuto nařízení, pokud
to přichází
v úvahu.
6.
Použití pro zdravotnické prostředky třídy IIa
V souladu s § 9 odst. 2 lze tuto přílohu použít na zdravotnické prostředky třídy
IIa s těmito odchylkami:
6.1. Odchylně od bodů 2., 3.1. a 3.2. na základě prohlášení o shodě výrobce zajišťuje
a prohlašuje, že jsou
zdravotnické prostředky třídy IIa vyrobeny ve shodě s technickou dokumentací podle
bodu 3.
přílohy č. 7 k tomuto nařízení a splňují požadavky tohoto nařízení, které se na ně
vztahují.
6.2. Pokud jde o zdravotnické prostředky třídy IIa, posoudí notifikovaná osoba jako
součást hodnocení
podle bodu 3.3. technickou dokumentaci podle bodu 3. přílohy č. 7 k tomuto nařízení
nejméně
1 reprezentativního vzorku z každé podskupiny zdravotnických prostředků s cílem posoudit
soulad
s ustanoveními tohoto nařízení.
6.3. Při volbě reprezentativního vzorku nebo vzorků bere notifikovaná osoba v úvahu
novost technologie,
podobnost návrhu, technologii, způsoby výroby a sterilizace, určené použití a výsledky
všech
předchozích souvisejících hodnocení (např. s ohledem na fyzikální, chemické a biologické
vlastnosti),
která byla provedena v souladu s tímto nařízením. Notifikovaná osoba zdokumentuje
a uchovává
pro potřebu příslušného orgánu státní správy důvody volby vzorku nebo vzorků.
6.4. Další vzorky posuzuje notifikovaná osoba jako součást dozoru podle bodu 4.3.
Příl.7
ES PROHLÁŠENÍ O SHODĚ
1.
ES prohlášení o shodě je postup, kterým výrobce nebo jeho zplnomocněný zástupce,
který plní povinnosti
podle bodu 2. a v případě zdravotnických prostředků uváděných na trh ve sterilním
stavu nebo
zdravotnických prostředků s měřicí funkcí povinnosti podle bodu 5., zajišťuje a prohlašuje,
že dotyčné
zdravotnické prostředky splňují požadavky tohoto nařízení, které se na ně vztahují.
2.
Výrobce musí vypracovat technickou dokumentaci popsanou v bodě 3. Výrobce zdravotnického
prostředku
nebo jeho zplnomocněný zástupce musí po dobu nejméně 5 let, a v případě implantabilních
zdravotnických prostředků nejméně po dobu 15 let, ode dne výroby posledního zdravotnického
prostředku
uchovávat pro potřebu příslušných orgánů státní správy technickou dokumentaci a prohlášení
o shodě.
3.
Technická dokumentace musí umožnit hodnocení shody
zdravotnického prostředku s požadavky tohoto nařízení; tato
dokumentace musí obsahovat zejména
3.1. celkový popis zdravotnického prostředku, včetně všech plánovaných variant, a
jeho určená použití,
3.2. konstrukční výkresy, připravované výrobní technologie
a schémata částí, podsestav, popřípadě obvodů,
3.3. popisy a vysvětlení nezbytné k porozumění výše uvedeným
výkresům, schématům a funkci zdravotnického prostředku,
3.4. výsledky analýzy rizik a seznam harmonizovaných norem,
které byly úplně nebo částečně použity, a popisy řešení
schválených pro splnění požadavků uvedených v § 4 odst. 1,
pokud harmonizované normy nebyly použity v plném
rozsahu,
3.5. v případě zdravotnických prostředků uváděných na trh ve sterilním stavu popis
použitých metod
a validační zprávu,
3.6. výsledky zejména konstrukčních výpočtů a provedených
kontrol; jestliže ke splnění určeného účelu použití má být
zdravotnický prostředek spojen s jiným zdravotnickým
prostředkem, popřípadě s jinými zdravotnickými prostředky,
musí být prokázáno, že zdravotnický prostředek vyhovuje
požadavkům uvedeným v § 4 odst. 1 při spojení se
zdravotnickým prostředkem nebo jinými zdravotnickými
prostředky, který má (mají) charakteristiky specifikované
výrobcem,
3.7. přijatá řešení, jak je uvedeno v části I. bodě 2. přílohy č. 1 k tomuto nařízení,
3.8. preklinické hodnocení,
3.9. klinické hodnocení podle přílohy č. 10 k tomuto nařízení,
3.10. etiketu nebo štítek zdravotnického prostředku a návod k jeho použití v českém
jazyce.
4.
Výrobce zavede a aktualizuje systematický postup vyhodnocování zkušeností získaných
s vyrobenými
zdravotnickými prostředky, včetně postupu podle ustanovení uvedených v příloze č.
10 k tomuto nařízení,
a vhodným způsobem zavádí nezbytná nápravná opatření. Součástí tohoto závazku je
povinnost
výrobce oznámit Ústavu nežádoucí příhody30) ihned, jakmile se o nich dozví.
5.
U zdravotnických prostředků uváděných na trh ve sterilním stavu
a u zdravotnických prostředků třídy I s měřicí funkcí výrobce
dodržuje kromě ustanovení této přílohy i jeden z postupů podle
příloh č. 2, 4, 5 nebo 6 k tomuto nařízení. Použití uvedených
příloh a opatření notifikované osoby se omezuje u zdravotnických prostředků
5.1. uváděných na trh ve sterilním stavu pouze na výrobní
hlediska, která se dotýkají zajišťování a udržování
sterilních podmínek,
5.2. s měřicí funkcí pouze na výrobní hlediska, která souvisejí
se shodou těchto prostředků s metrologickými požadavky,
přičemž lze použít ustanovení bodu 6.1. této přílohy.
6.
Použití pro prostředky třídy IIa.
6.1. V souladu s § 9 odst. 2 lze tuto přílohu použít pro
zdravotnické prostředky třídy IIa s tím, že
6.1.1. při použití této přílohy ve spojení s postupem
podle příloh č. 4, 5 nebo 6 k tomuto nařízení tvoří
prohlášení o shodě podle uvedených příloh jediné
prohlášení o shodě. V tomto prohlášení, které
vychází z této přílohy, výrobce zaručuje
a prohlašuje, že návrh zdravotnického prostředku
vyhovuje ustanovením, která se na něj vztahují
z tohoto nařízení.
Příl.8
PROHLÁŠENÍ O ZDRAVOTNICKÝCH PROSTŘEDCÍCH PRO ZVLÁŠTNÍ ÚČELY
1.
Výrobce nebo jeho zplnomocněný zástupce vyhotoví o zakázkových
zdravotnických prostředcích nebo o zdravotnických prostředcích
určených pro klinické zkoušky prohlášení.
2.
Prohlášení uvedené v bodě 1 této přílohy musí obsahovat
2.1. u zakázkových zdravotnických prostředků
2.1.1. jméno, popřípadě jména, a příjmení výrobce, adresu jeho bydliště a místa podnikání,
jestliže
výrobcem je fyzická osoba žijící v zahraničí, jméno, popřípadě jména, a příjmení,
adresu
trvalého pobytu výrobce, jestliže má trvalý pobyt v České republice; název nebo obchodní
firmu, adresu sídla, jestliže výrobcem je právnická osoba.
2.1.2. údaje umožňující identifikaci příslušného
zdravotnického prostředku,
2.1.3. prohlášení, že tento zdravotnický prostředek je
určen výlučně pro určitého pacienta spolu
s uvedením jeho příjmení,
2.1.4. jméno, popřípadě jména a příjmení lékaře nebo jiné
pověřené osoby, která vypracovala předpis tohoto
zdravotnického prostředku a název nebo obchodní
firmu osoby oprávněné poskytovat zdravotní péči
podle zvláštních právních předpisů,31)
2.1.5. specifické vlastnosti zdravotnického prostředku, jak jsou uvedeny v
předpisu zdravotnického
prostředku,
2.1.6. prohlášení, že tento zdravotnický prostředek
vyhovuje základním požadavkům, popřípadě údaje,
které z těchto základních požadavků nejsou zcela
splněny včetně odůvodnění,
2.2. u zdravotnických prostředků určených ke klinickým zkouškám
podle zákona o zdravotnických prostředcích a přílohy č.
10 k tomuto nařízení
2.2.1. údaje umožňující identifikaci tohoto prostředku,
2.2.2. plán klinických zkoušek,
2.2.3. soubor informací pro zkoušejícího,
2.2.4. potvrzení o pojištění subjektů,
2.2.5. dokumenty použité k získání informovaného souhlasu,
2.2.6. prohlášení, zda zdravotnický prostředek obsahuje jako svou integrální součást
látku nebo
derivát z lidské krve uvedené v bodě 7.4. přílohy č. 1 k tomuto nařízení,
2.2.7. prohlášení, zda je zdravotnický prostředek vyroben s použitím tkání zvířecího
původu ve
smyslu § 1 písm. b),
2.2.8. stanovisko příslušné etické komise včetně
podrobných údajů spojených s tímto stanoviskem,
2.2.9. název právnické osoby nebo jméno, popřípadě jména a příjmení fyzické osoby,
která zadává
provedení klinické zkoušky, a jméno, popřípadě jména a příjmení lékaře nebo jiné
pověřené osoby, která prakticky provádí klinickou zkoušku; na tyto osoby se vztahují
povinnosti a odpovědnost uvedené v § 8 až 14 zákona o zdravotnických prostředcích,
2.2.10. místo, datum zahájení a plánovaná doba trvání
klinických zkoušek,
2.2.11. prohlášení, že příslušný zdravotnický prostředek
vyhovuje základním požadavkům, s výjimkou hledisek,
které jsou předmětem klinických zkoušek a že
s ohledem na tato hlediska byla učiněna předběžná
opatření k ochraně zdraví a bezpečnosti pacienta.
3.
Výrobce zpřístupňuje příslušným úřadům státní správy
3.1. u zakázkových zdravotnických prostředků dokumentaci, která uvádí výrobní místo
nebo místa
a která umožní pochopení návrhu, výroby a funkční způsobilosti zdravotnického prostředku,
včetně očekávané funkční způsobilosti, aby bylo možné posoudit shodu s požadavky
tohoto nařízení;
přitom výrobce učiní nezbytná opatření, aby jeho výrobní proces produkoval zakázkové
zdravotnické prostředky v souladu s uvedenou dokumentací,
3.2. u zdravotnických prostředků určených ke klinickým zkouškám dokumentace obsahuje
3.2.1. celkový popis zdravotnického prostředku a jeho určená použití,
3.2.2. konstrukční výkresy, předpokládané výrobní technologie, zejména pokud jde
o sterilizaci,
a schémata součástí, podsestav, obvodů atd.,
3.2.3. popisy a vysvětlivky potřebné pro pochopení výše uvedených výkresů, schémat
a fungování
zdravotnického prostředku,
3.2.4. výsledky analýzy rizik a seznam norem uvedených v § 4 odst. 2, které byly
zcela nebo
částečně použity, a popis řešení přijatých pro splnění základních požadavků tohoto
nařízení,
pokud nebyly použity normy podle § 4 odst. 2,
3.2.5. pokud zdravotnický prostředek obsahuje jako svou integrální součást látku
nebo derivát
z lidské krve uvedené v bodě 7.4. přílohy č. 1 k tomuto nařízení, údaje o zkouškách
provedených
v této souvislosti požadovaných k posouzení bezpečnosti, jakosti a užitečnosti této
látky nebo derivátu z lidské krve s přihlédnutím k určenému účelu použití zdravotnického
prostředku,
3.2.6. pokud je zdravotnický prostředek vyroben s použitím tkání zvířecího původu
ve smyslu § 1
písm. b), opatření řízení rizik v této souvislosti, která byla uplatněna za účelem
snížení rizika
přenosu nákazy,
3.2.7. výsledky provedených konstrukčních výpočtů, kontrol, technických zkoušek atd.
Přitom výrobce musí přijmout veškerá opatření nezbytná k zajištění toho, že výsledkem
výrobního
postupu jsou zdravotnické prostředky, které jsou vyrobeny v souladu s dokumentací
uvedenou
v bodech 3.2.1. až 3.2.7.
Dále výrobce musí povolit posouzení nebo případně audit účinnosti těchto opatření.
4.
Informace obsažené v prohlášeních podle této přílohy se uchovávají po dobu nejméně
5 let a v případě
implantabilních zdravotnických prostředků po dobu nejméně 15 let.
5.
U zakázkových zdravotnických prostředků výrobce vyhodnocuje a dokumentuje zkušenosti
získané
s vyrobenými zdravotnickými prostředky, včetně ustanovení uvedených v příloze č.
10 k tomuto nařízení,
a vhodným způsobem provádí nezbytná nápravná opatření. Součástí této povinnosti je
oznamování
Ústavu nežádoucí příhody30) výrobcem, jakmile se o ní dozví, a rovněž oznamování
souvisejících
nápravných opatření.
Příl.9
KLASIFIKAČNÍ PRAVIDLA
I.
VYSVĚTLIVKY
1.
Seznam vysvětlivek pro klasifikační pravidla:
1.1. trvání
1.1.1. "trvání přechodné" znamená nepřetržité použití
zdravotnického prostředku po dobu kratší než 60
minut,
1.1.2. "trvání krátkodobé" znamená nepřetržité použití
zdravotnického prostředku po dobu kratší než 30
dnů,
1.1.3. "trvání dlouhodobé" znamená nepřetržité použití
zdravotnického prostředku po dobu delší než 30 dnů,
1.2. invazivní prostředky
1.2.1. "tělní otvor" znamená přirozený otvor v těle,
včetně vnějšího povrchu oční bulvy nebo trvalý,
uměle vytvořený otvor,
1.2.2. "invazivní zdravotnický prostředek" znamená
zdravotnický prostředek, který zcela nebo zčásti
proniká do těla tělním otvorem nebo povrchem těla,
1.2.3. "chirurgicky invazivní zdravotnický prostředek"
znamená invazivní zdravotnický prostředek, který
proniká do těla jeho povrchem pomocí chirurgického
zásahu nebo v souvislosti s ním.
Pro účely tohoto nařízení se na zdravotnické
prostředky, které nejsou vymezeny podle předchozí
věty a které pronikají do těla jinak, než
vytvořeným otvorem, pohlíží jako na chirurgicky
invazivní zdravotnické prostředky,
1.2.4. "implantabilní zdravotnický prostředek" znamená
zdravotnický prostředek, který má být zcela zaveden
do lidského těla nebo má nahradit epiteliální
povrch nebo povrch oka chirurgickým zákrokem, po
němž má zůstat na místě; za implantabilní
zdravotnický prostředek se rovněž považuje
zdravotnický prostředek, který má být chirurgickým
zákrokem částečně zaveden do lidského těla a po
zákroku v něm zůstat alespoň 30 dní,
1.3. "chirurgický nástroj pro opakované použití" znamená
nástroj určený k chirurgickému řezání, vrtání, řezání
pilkou, škrabání, seškrabávání, svorkování, odtahování,
spínání a podobným postupům, který není spojen s aktivním
zdravotnickým prostředkem a může být po provedení
příslušných postupů znovu použit,
1.4. „aktivní zdravotnický prostředek“ znamená zdravotnický prostředek, jehož činnost
závisí na
zdroji elektrické nebo jiné energie, která není přímo vytvářena lidským tělem nebo
gravitací a který
působí prostřednictvím přeměny této energie; zdravotnické prostředky určené k přenosu
energie
nebo látek mezi aktivním zdravotnickým prostředkem a pacientem bez jakékoliv významné
změny
se za aktivní zdravotnický prostředek nepovažují; samostatné programové vybavení
se považuje za
aktivní zdravotnický prostředek,
1.5. "aktivní terapeutické zařízení" znamená aktivní
zdravotnický prostředek použitý samostatně nebo
v kombinaci s dalšími zdravotnickými prostředky
k podpoře, změně, náhradě, úpravě nebo obnovení
biologických funkcí nebo struktur za účelem léčby nebo
zmírnění nemoci, poranění nebo zdravotního postižení,
1.6. "aktivní diagnostický zdravotnický prostředek" znamená
aktivní zdravotnický prostředek použitý samostatně nebo
v kombinaci s dalšími zdravotnickými prostředky
k dodávání informací pro diagnostikování, monitorování,
zjišťování nebo léčbu fyziologických stavů, stavu zdraví,
nemocí nebo vrozených vad,
1.7. „Centrální oběhový systém“ znamená pro účely tohoto nařízení tyto cévy: arteriae
pulmonales,
aorta ascendens, arcus aortae, aorta descendens do bifurcatio aortae, arteriae coronariae,
arteria
carotis communis, arteria carotis externa, arteria carotis interna, arteriae cerebrales,
truncus brachiocephalicus,
venae cordis, venae pulmonales, vena cava superior, vena cava inferior.
1.8. "centrální nervový systém" znamená pro účely tohoto
nařízení mozek, mozkové pleny a páteřní míchu,
1.9. "endoprotézou kyčle, endoprotézou kolena a endoprotézou ramena"
se pro účely tohoto nařízení rozumí implantabilní součásti systému
totální endoprotézy, jejichž úkolem je zajistit funkci podobnou přirozenému
kyčelnímu kloubu, přirozenému kolennímu kloubu a přirozenému ramennímu
kloubu. Toto vymezení se nevztahuje na pomocné součásti (šrouby, klíny,
dlahy či nástroje).
II.
PROVÁDĚCÍ PRAVIDLA
2.
Prováděcí pravidla.
2.1. Klasifikační pravidla se řídí určeným účelem použití
zdravotnických prostředků.
2.2. Jestliže je zdravotnický prostředek určen k použití
v kombinaci s jiným zdravotnickým prostředkem, uplatní se
klasifikační pravidla pro každý zdravotnický prostředek
samostatně.
Příslušenství se klasifikuje samostatně (odděleně od
zdravotnického prostředku, se kterým se používá).
2.3. Programové vybavení, které řídí nebo ovlivňuje použití
zdravotnického prostředku, patří do stejné třídy jako
tento zdravotnický prostředek.
2.4. Není-li zdravotnický prostředek určen výhradně nebo
z principu k použití pro určitou část těla, posuzuje se
a je klasifikován podle zásady nejkritičtějšího určeného
účelu použití.
2.5. Platí-li pro klasifikaci zdravotnického prostředku několik
pravidel vycházejících z účinnosti tohoto prostředku
a určeného účelu použití stanovených výrobcem, použijí se
nejpřísnější pravidla ve vyšší klasifikační třídě.
2.6. Při výpočtu doby uvedené v bodě 1.1. části I. se nepřetržitým použitím rozumí
skutečné nepřerušené
použití zdravotnického prostředku pro určený účel. Pokud je však použití zdravotnického
prostředku přerušeno za účelem okamžité výměny zdravotnického prostředku za stejný
nebo
totožný zdravotnický prostředek, je to považováno za pokračování v nepřetržitém použití
zdravotnického
prostředku.
III.
KLASIFIKACE
Pro klasifikaci druhů zdravotnických prostředků se používají dále
uvedená pravidla:
1.
Neinvazivní zdravotnické prostředky.
1.1. Pravidlo 1.
Neinvazivní zdravotnické prostředky patří do třídy I,
pokud se na ně nevztahuje některé z dalších pravidel.
1.2. Pravidlo 2.
Neinvazivní zdravotnické prostředky určené pro odvádění
nebo shromažďování krve, tělních tekutin nebo tkání,
tekutin nebo plynů pro případnou infúzi, podávání nebo
zavádění do těla, patří do třídy IIa, jestliže
1.1.2. mohou být připojeny k aktivnímu zdravotnickému
prostředku třídy IIa nebo vyšší třídy,
1.2.2. jsou určeny pro odvádění nebo shromažďování krve
nebo jiných tělních tekutin nebo pro uchovávání
orgánů, částí orgánů nebo tělesných tkání.
Ve všech ostatních případech patří do třídy I.
1.3. Pravidlo 3.
Neinvazivní zdravotnické prostředky určené pro měnění
biologického nebo chemického složení krve, jiných tělních
tekutin nebo jiných tekutin určených pro infúzi do těla
patří do třídy IIb; pokud však léčba spočívá ve filtraci,
odstředění či výměně plynu nebo tepla, patří do třídy IIa.
1.4. Pravidlo 4.
Neinvazivní zdravotnické prostředky, které přicházejí do
styku s poraněnou kůží patří do
1.4.1. třídy I, jestliže jsou určeny k použití jako
mechanická překážka, ke kompresi nebo k absorpci
výpotků,
1.4.2. třídy IIb, jestliže jsou určeny z principu
k použití u ran, při kterých byla porušena dermis
a mohou mít pouze sekundární terapeutický účinek,
1.4.3. třídy IIa v ostatních případech, včetně
zdravotnických prostředků z principu určených
k ošetření mikroprostředí rány.
2.
Invazivní zdravotnické prostředky.
2.1. Pravidlo 5.
Všechny invazivní zdravotnické prostředky
k použití pro přirozené tělesné otvory jiné než
chirurgicky invazivní zdravotnické prostředky a neurčené
pro napojení na aktivní zdravotnický prostředek
nebo určené pro napojení na aktivní zdravotnický prostředek
třídy I, patří do
2.1.1. třídy I, jestliže jsou určeny k přechodnému
použití,
2.1.2. třídy IIa, jestliže jsou určeny ke krátkodobému
použití, s výjimkou použití v ústní dutině až po
hltan, ve zvukovodu až po ušní bubínek nebo
v nosní dutině, kdy patří do třídy I,
2.1.3. třídy IIb, jestliže jsou určeny k dlouhodobému
použití, s výjimkou použití v ústní dutině až po
hltan, ve zvukovodu až po ušní bubínek nebo
v nosní dutině a není pravděpodobná jejich absorpce
sliznicí, kdy patří do třídy IIa.
Invazivní zdravotnické prostředky, které se vztahují
k tělním otvorům a které nejsou chirurgicky invazivními
zdravotnickými prostředky a jsou určené ke spojení
s aktivním zdravotnickým prostředkem třídy IIa nebo vyšší,
patří do třídy IIa.
2.2. Pravidlo 6.
Všechny chirurgicky invazivní zdravotnické prostředky pro přechodné použití spadají
do třídy IIa,
pokud nejsou
a) zvlášť určeny pro kontrolu, diagnostiku, sledování nebo nápravu vad srdce nebo
centrálního
oběhového systému přímým kontaktem s těmito částmi těla, kdy spadají do třídy III,
b) chirurgickými nástroji pro opakované použití, kdy spadají do třídy I,
c) zvlášť určeny pro přímý kontakt s centrální nervovou soustavou, kdy spadají do
třídy III,
d) určeny k dodávání energie ve formě ionizujícího záření, kdy spadají do třídy IIb,
e) určeny k vyvolání biologického účinku nebo k úplné či převážné absorpci, kdy spadají
do třídy
IIb,
f) určeny k podávání léčivých přípravků dávkovacím systémem, pokud se tak děje způsobem
potenciálně nebezpečným z hlediska způsobu aplikace, kdy spadají do třídy IIb.
2.3. Pravidlo 7.
Chirurgicky invazivní zdravotnické prostředky pro
krátkodobé použití patří do třídy IIa, jestliže nejsou
určeny
2.3.1. zvlášť pro kontrolu, diagnostiku, sledování nebo korekci srdeční vady nebo
vady centrálního
oběhového systému přímým kontaktem s těmito částmi těla, kdy spadají do třídy III,
2.3.2. specificky pro použití v přímém dotyku
s centrálním nervovým systémem, kdy patří do třídy
III,
2.3.3. k dodávání energie ve formě ionizujícího záření,
kdy patří do třídy IIb,
2.3.4. k vyvolání biologického účinku nebo k částečné či
plné absorpci, kdy patří do třídy III,
2.3.5. k uskutečnění chemické změny v těle, s výjimkou
zdravotnických prostředků umístěných v zubech, nebo
k podávání léčiv, kdy patří do třídy IIb.
2.4. Pravidlo 8.
Implantabilní a dlouhodobě chirurgicky invazivní
zdravotnické prostředky patří do třídy IIb, jestliže
nejsou určeny k
2.4.1. umístění v zubech, kdy patří do třídy IIa,
2.4.2. použití v přímém dotyku se srdcem, s centrálním
oběhovým nebo centrálním nervovým systémem, kdy
patří do třídy III,
2.4.3. vyvolání biologického účinku nebo k částečné či
plné absorpci, kdy patří do třídy III,
2.4.4. uskutečnění chemické změny v těle s výjimkou
zdravotnických prostředků umístěných v zubech, nebo
k podávání léčiv, kdy patří do třídy III.
3.
Dodatečná pravidla pro aktivní zdravotnické prostředky.
3.1. Pravidlo 9.
3.1.1. Aktivní terapeutické zdravotnické prostředky určené
k podávání nebo výměně energie patří do třídy IIa,
jestliže nejsou jejich charakteristiky takové, že
s přihlédnutím k povaze, hustotě a místu aplikace
energie mohou energii do lidského těla nebo z těla
předávat nebo vyměňovat potenciálně nebezpečným
způsobem, kdy patří do třídy IIb.
3.1.2. Aktivní zdravotnické prostředky určené k řízení
nebo monitorování účinnosti aktivních
terapeutických zdravotnických prostředků třídy IIb
nebo určené přímo k ovlivňování účinnosti takových
zdravotnických prostředků patří do třídy IIb.
3.2. Pravidlo 10.
Aktivní zdravotnické prostředky určené pro diagnostiku
patří do třídy IIa, jestliže jsou určeny k
3.2.1. podávání energie, která je lidským tělem
absorbována, s výjimkou zdravotnických prostředků
používaných k osvětlení pacientova těla ve
viditelném spektru,
3.2.2. zobrazení in vivo distribuce radiofarmak,
3.2.3. přímé diagnostice nebo monitorování životně
důležitých fyziologických procesů, pokud nejsou
specificky určeny k monitorování životně důležitých
fyziologických parametrů, kde povaha změn je
taková, že by mohlo dojít k bezprostřednímu
ohrožení pacienta (například změny srdečního
výkonu, dýchání, činnosti centrální nervové
soustavy), kdy patří do třídy IIb.
Aktivní zdravotnické prostředky určené k emitování
ionizujícího záření a určené pro diagnostickou
a terapeutickou intervenční radiologii, včetně
zdravotnických prostředků, které řídí nebo
monitorují takové zdravotnické prostředky či přímo
ovlivňují jejich účinnost, patří do třídy IIb.
3.3. Pravidlo 11.
Aktivní zdravotnické prostředky určené k podávání léčiv,
tělních tekutin nebo jiných látek do těla nebo
odstraňování léčiv, tělních tekutin nebo jiných látek
z těla patří do třídy IIa, pokud se tak nečiní způsobem,
který je potenciálně nebezpečný s přihlédnutím k povaze
používaných látek, částí těla a ke způsobu aplikace, kdy
patří do třídy IIb.
3.4. Pravidlo 12.
Ostatní aktivní zdravotnické prostředky patří do třídy I.
4.
Zvláštní pravidla.
4.1. Pravidlo 13.
4.1.1. Zdravotnické prostředky obsahující jako integrální
část látku, která při samostatném použití může být
považována za léčivo a která může působit na
lidské tělo účinkem doplňujícím účinek
zdravotnického prostředku, patří do třídy III.
4.1.2. Zdravotnické prostředky, které obsahují jako svou
integrální součást derivát z lidské krve patří do
třídy III.
4.2. Pravidlo 14.
Zdravotnické prostředky používané pro antikoncepci nebo
k prevenci přenosu sexuálně přenosných chorob patří do
třídy IIb, jestliže nejsou implantabilními nebo dlouhodobě
invazivními zdravotnickými prostředky, kdy patří do třídy
III.
4.3. Pravidlo 15.
Zdravotnické prostředky specificky určené k použití při
dezinfekci, čištění, oplachování, popřípadě hydrataci
kontaktních čoček, patří do třídy IIb.
Zdravotnické prostředky specificky určené k použití při
dezinfekci zdravotnických prostředků patří do třídy IIa,
pokud nejsou zvlášť určeny pro dezinfekci invazivních
zdravotnických prostředků, kdy patří do třídy IIb.
Toto pravidlo neplatí pro výrobky určené k fyzickému
čištění zdravotnických prostředků, které nejsou
kontaktními čočkami.
4.4. Pravidlo 16.
Zdravotnické prostředky specificky určené pro
záznam diagnostických rentgenových zobrazení patří do
třídy IIa.
4.5. Pravidlo 17.
Zdravotnické prostředky vyrobené s použitím zvířecích
tkání nebo jejich neživých derivátů patří do třídy III,
s výjimkou zdravotnických prostředků, které jsou určeny
pouze ke styku s neporušenou kůží.
5.
Pravidlo 18.
Odchylně od jiných pravidel patří krevní vaky do třídy IIb.
6.
Pravidlo 19.
Odchylně od jiných pravidel patří prsní implantáty do třídy
III.
7.
Pravidlo 20.
Odchylně od jiných pravidel patří endoprotézy kyčle, endoprotézy
kolena a endoprotézy ramena do třídy III.
Příl.10
BLIŽŠÍ INFORMACE KE KLINICKÉMU HODNOCENÍ
1.
Obecná ustanovení
1.1. Obecně platí, že potvrzení shody s požadavky, které se týkají vlastností a funkční
způsobilosti podle
bodů 1. a 3. přílohy č. 1 k tomuto nařízení za běžných podmínek použití zdravotnického
prostředku,
a hodnocení vedlejších účinků a přijatelnosti rizika uvedené v bodě 6. části I. přílohy
č. 1
k tomuto nařízení musí být založeny na klinických údajích. Vyhodnocení těchto údajů
(dále jen
„klinické hodnocení“), případně s přihlédnutím k veškerým příslušným harmonizovaným
normám,
vyplývá z definovaného a metodologicky platného postupu založeného
1.1.1. buď na kritickém hodnocení v současné době dostupné související odborné literatury,
která se
vztahuje k bezpečnosti, funkční způsobilosti, vlastnostem návrhu a určenému účelu
použití
zdravotnického prostředku, jestliže
1.1.1.1. existuje důkaz rovnocennosti zdravotnického prostředku se zdravotnickým
prostředkem,
kterého se údaje týkají, a
1.1.1.2. údaje náležitě dokazují shodu s příslušnými základními požadavky;
1.2. V případě implantabilních zdravotnických prostředků a zdravotnických prostředků
třídy III se
klinické zkoušky provádějí vždy, kromě řádně odůvodněných případů, kdy se lze spolehnout
na
existující klinické údaje.
1.3. Klinické hodnocení a jeho výstupy se dokumentují. Tato dokumentace klinického
hodnocení je
součástí technické dokumentace zdravotnického prostředku nebo musí být v technické
dokumentaci
plné odkazy.
1.4. Klinické hodnocení a dokumentace, která se k němu vztahuje, musí být aktivně
aktualizovány
s použitím údajů z dozoru po uvedení zdravotnických prostředků na trh. Pokud není
následné
klinické sledování po uvedení na trh jako součást plánu dozoru po uvedení zdravotnických
prostředků
na trh považováno za nezbytné, musí to být náležitě odůvodněno a zdokumentováno.
1.5. Není-li prokázání shody se základními požadavky založené na klinických údajích
považováno za
vhodné, musí být každé takové vyloučení náležitě odůvodněno na základě řízení rizika
a s ohledem
na zvláštní povahu interakce zdravotnického prostředku a těla, na zamýšlené klinické
výkony a na
tvrzení výrobce. Dostatečnost prokázání shody se základními požadavky pouze hodnocením
výkonu,
srovnávacím testováním a preklinickým hodnocením musí být řádně odůvodněna.“.
1.6. Všechny tyto údaje se považují v souladu s ustanovením
§ 49 zákona o zdravotnických prostředcích za důvěrné.
2.
Klinické zkoušky
2.1. Účel
Účelem klinických zkoušek je ověřit, zda je funkční způsobilost zdravotnických prostředků
za
běžných podmínek použití v souladu s požadavky podle bodu 3. přílohy č. 1 k tomuto
nařízení,
a určit všechny nežádoucí vedlejší účinky za běžných podmínek použití a posoudit,
zda představují
s ohledem na určenou funkční způsobilost prostředku přijatelná rizika.
2.2. Etická hlediska
Klinické zkoušky musí být prováděny v souladu s Helsinskou deklarací, přijatou 18.
Světovým
zdravotnickým shromážděním v roce 1964 v Helsinkách ve Finsku a naposledy pozměněnou
Světovým
zdravotnickým shromážděním. Je podstatné, že všechna opatření pro ochranu osob musí
být
přijímána v duchu Helsinské deklarace. Tento požadavek se vztahuje na klinické zkoušky,
a to od
první úvahy o jejich potřebě a oprávněnosti až po zveřejnění výsledků.
2.3. Metody
2.3.1. Klinické zkoušky se provádějí na základě vhodného zkušebního plánu, který
odráží nejnovější
stav vědeckých a technických znalostí a je koncipován tak, aby potvrdil nebo vyvrátil
odůvodněné očekávání výrobce ve vztahu k prostředku. Zkoušky musí obsahovat příslušný
počet pozorování, aby byla zaručena vědecká platnost závěrů.
2.3.2. Postupy použité při zkouškách musí být pro zkoušený prostředek přiměřené.
2.3.3. Klinické zkoušky se provádějí za podmínek podobných běžným podmínkám použití
prostředku.
2.3.4. Zkoušejí se všechny příslušné charakteristiky prostředku, včetně těch, které
se týkají bezpečnosti
a funkční způsobilosti prostředku a jeho vlivu na pacienty.
2.3.5. Veškeré závažné nežádoucí příhody musí být plně zaznamenány a neprodleně oznámeny
všem příslušným orgánům členských států, v nichž probíhají klinické
zkoušky.
2.3.6. Zkoušky se provádějí na odpovědnost zkoušejícího nebo jiné, kvalifikací k
tomu oprávněné
osoby, a ve vhodném prostředí. Zkoušejícímu nebo jiné oprávněné osobě musí být zpřístupněny
technické a klinické údaje týkající se prostředku.
2.3.7. Písemná zpráva, podepsaná zkoušejícím nebo jinou oprávněnou osobou, obsahuje
kritické
hodnocení všech údajů shromážděných během klinických zkoušek.
3.
Po ukončení klinických zkoušek se zpracovává závěrečná písemná zpráva v souladu
s § 11 písm. c) zákona o zdravotnických prostředcích.
Příl.11
KRITÉRIA, KTERÁ MUSÍ SPLŇOVAT NOTIFIKOVANÉ OSOBY
Úřad uděluje autorizaci za předpokladu splnění následujících
podmínek a požadavků
1.
notifikovaná osoba a fyzické osoby, které jsou jejím
statutárním orgánem nebo jeho členy, a zaměstnanci notifikované
osoby, kteří jsou zodpovědní za hodnotící a ověřovací činnosti
uvedené v přílohách č. 2 až 6 k tomuto nařízení (dále jen
"hodnocení a ověřování"), nesmějí
1.1. být autorem návrhu zdravotnického prostředku, jeho
výrobcem, distributorem ani osobou provádějící údržbu,
opravy, seřizování a kontroly zdravotnických prostředků
nebo poskytovatelem32) těchto prostředků,
1.2. být zplnomocněným zástupcem osoby uvedené v bodu 1,
1.3. podílet se přímo na návrhu, konstrukci, prodeji
a udržování zdravotnických prostředků ani zastupovat
strany zúčastněné na těchto činnostech;
možnost výměny technických informací mezi výrobcem
a notifikovanou osobou není předchozí částí věty dotčena,
2.
notifikovaná osoba a její zaměstnanci provádějí hodnocení
a ověřování na soudobé úrovni vědy a techniky, s odpovídající
odborností a způsobilostí v oblasti zdravotnických prostředků,
zodpovědností, objektivitou a za okolností, které nemohou
ovlivnit jejich úsudek nebo výsledky kontrolní činnosti,
zejména které vylučují vliv osob, majících zájem na výsledcích
hodnocení a ověřování,
3.
notifikovaná osoba
3.1. musí být schopná vykonat úkoly, které jsou jí určeny podle
jedné z příloh č. 2 až 6 k tomuto nařízení a vykonat další
úkoly, pro které byla notifikována, a to buď sama nebo
smluvně je nechat provést na svou zodpovědnost osobou,
kterou za tímto účelem sjedná. Předpokladem je, že
notifikovaná osoba bude mít k dispozici dostatečné vědecké
personální zázemí s dostatečnými zkušenostmi a znalostmi
potřebnými k posouzení bezpečnosti, účinnosti a vhodnosti
zdravotnických prostředků pro používání při poskytování
zdravotní péče, pro které byla notifikována, s ohledem na
požadavky vyplývající z tohoto nařízení, zejména
z přílohy č. 1 k tomuto nařízení,
3.2. musí mít přístup k vybavení nezbytnému k provedení
požadovaného ověření,
3.3. při smluvním zajištění úkolů spojených s hodnocením
a ověřováním se musí nejprve ujistit, že sjednaná jiná
osoba vyhovuje ustanovením tohoto nařízení zejména této
přílohy; pro potřebu příslušných úřadů státní správy
uchovává dokumenty o kvalifikaci sjednané osoby a údaje
o její činnosti vykonané podle tohoto nařízení,
4.
notifikovaná osoba musí
4.1. být řádně odborně zapracována pro všechny hodnotící
a ověřovací postupy, pro které je ustanovena,
4.2. být obeznámena s kontrolní činností, kterou provádí a musí
mít odpovídající zkušenosti s těmito činnostmi,
4.3. být schopna vypracovat příslušné certifikáty, záznamy
a zprávy prokazující, že kontroly byly provedeny,
4.4. zaručovat nestrannost hodnocení a ověřování,
5.
nezávislost notifikované osoby musí být zaručena; její odměny
(platy) nesmějí být závislé na počtu provedených kontrol ani na
jejich výsledcích,
6.
notifikovaná osoba je povinna v souladu se zvláštním právním
předpisem uzavřít smlouvu o pojištění odpovědnosti za škodu,33)
7.
povinnost osob uvedených v návětí v bodu 1 zachovávat
mlčenlivost o skutečnostech, o nichž se dozvídají při činnosti
notifikované osoby stanoví zvláštní právní předpis.34)
Příl.12
1.
ANALÝZA RIZIK A ŘÍZENÍ RIZIK
1.1. Výrobce pro zdravotnické prostředky uvedené v § 1 písm. b)
1.1.1. provádí analýzu rizik a řízení rizik,
1.1.2. odůvodňuje své rozhodnutí použít předmětné tkáně
nebo deriváty zvířecího původu k výrobě těchto
zdravotnických prostředků s tím, že zevrubně uvede
druhy zvířat a tkání, které mají být použity, a
1.1.3. zohledňuje očekávaný klinický přínos, potenciální
zbytkové riziko a vhodná náhradní řešení.
1.2. Postup posouzení
Výrobce za účelem zajištění vysoké úrovně ochrany pacientů
nebo uživatelů
1.2.1. užívá u zdravotnických prostředků vyrobených za
použití tkání nebo derivátů zvířecího původu podle
bodu 1.1.2. vhodných a dobře dokumentovaných
strategií analýzy rizik a řízení rizik, přičemž
vezme v úvahu všechny významné aspekty týkající se
TSE,
1.2.2. identifikuje nebezpečí spojené s dotyčnými tkáněmi
nebo deriváty,
1.2.3. zakládá dokumentaci o opatřeních přijatých
k minimalizaci rizika přenosu a k prokázání
přijatelnosti zbytkových rizik spojených se
zdravotnickým prostředkem vyrobeným za použití
takové tkáně nebo derivátu,
1.2.4. zohledňuje určený účel použití,35)
1.2.5. analyzuje, hodnotí a zpracovává všechny faktory
uvedené v bodech 1.2.6. až 1.2.14. této přílohy, na
kombinaci těchto opatření závisí bezpečnost
zdravotnického prostředku ve smyslu jeho potenciálu
přenosu přenosných původců; zejména má na zřeteli
1.2.5.1. výběr výchozích materiálů (tkání nebo
derivátů, které jsou považovány za vhodné
vzhledem k jejich potenciální kontaminaci
přenosnými původci), přičemž se bere
v úvahu jejich další zpracování,
1.2.5.2. uplatnění výrobních postupů k odstranění
přenosných původců z tkání nebo derivátů
z kontrolovaných zdrojů nebo k jejich
deaktivování,
1.2.5.4. stanoviska přijatá příslušnými vědeckými
výbory, popřípadě stanoviska Výboru pro
hromadně vyráběné léčivé přípravky
publikovaná v Úředním věstníku Evropské
unie,
1.2.5.5. nutnost zohlednění vlastností
zdravotnického prostředku a jeho určený
účel použití.
1.2.6. Zvířata jako zdroj materiálu
Riziko TSE závisí na druhu a plemenu (rase) zvířat
a na povaze výchozí tkáně. Protože se infekčnost
TSE během inkubační doby několika let akumuluje,
považuje se použití mladých zdravých zvířat jako
zdroje materiálu za faktor snižující riziko.
Riziková zvířata jako zvířata uhynulá nebo nuceně
utracená; zvířata s podezřením na TSE musí být
vyloučena.
1.2.7. Geografický původ
1.2.7.1. Až do vytvoření klasifikace zemí na
základě stavu BSE podle práva Evropské
unie,36) se při posuzování rizika
země původu použije geografické riziko BSE
(Geographical BSE Risk), které se považuje
za kvalitativní indikátor pravděpodobnosti
výskytu jednoho či více kusů skotu
infikovaných BSE, pre-klinicky i klinicky,
v určitém časovém bodě v dané zemi. Stupeň
infekce, tam, kde je výskyt potvrzen, se
udává podle následující tabulky.
----------------------------------------------------------
Stupeň Výskyt jednoho či více kusů skotu klinicky či
pre-klinicky infikovaných původcem BSE, na
geografickém území / v zemi
----------------------------------------------------------
I Vysoce nepravděpodobný
----------------------------------------------------------
II Nepravděpodobný, avšak nikoli vyloučený
----------------------------------------------------------
III Pravděpodobný, ale nepotvrzený nebo potvrzený na
nižším stupni
----------------------------------------------------------
IV Potvrzený, na vyšším stupni
----------------------------------------------------------
1.2.7.2. Určité faktory ovlivňují geografické
riziko infekce BSE spojené s používáním
surových tkání nebo derivátů
z jednotlivých zemí. Tyto faktory jsou
definovány v mezinárodním dokumentu.37)
1.2.7.3. Vědecký řídící výbor provádí posouzení
geografického rizika BSE v členských státech
a v některých třetích zemích; toto
posouzení bude zahrnovat všechny země,
které požádaly o kategorizaci stavu BSE
při zohlednění hlavních faktorů uvedených
v mezinárodním dokumentu.
1.2.8. Povaha výchozí tkáně
Výrobce
1.2.8.1. bere v úvahu klasifikaci nebezpečí
vztahujících se k různým typům výchozí
tkáně; výběr tkáně zvířecího původu musí
podléhat kontrole a individuální
veterinární inspekci, poražená zvířata
musí být označena jako vhodná pro lidskou
spotřebu,
1.2.8.2. zajistí vyloučení jakéhokoli rizika
křížené kontaminace během porážky zvířete,
1.2.8.3. nesmí použít žádné tkáně nebo deriváty
zvířecího původu klasifikované jako
potenciálně vysoce TSE-infekční, pokud
není použití těchto materiálů ve
výjimečných případech nezbytné,
1.2.8.4. zohlední významné přínosy pro pacienta
a neexistenci jakékoli alternativní
výchozí tkáně,
1.2.8.5. použije ustanovení příslušných předpisů
Evropské unie.38)
1.2.9. Ovce a kozy
Podle současných znalostí na základě titrů
přenosných původců v tkáních, tělních tekutinách
přirozeně infikovaných ovcí a koz s klinickou
klusavkou (scrapie) byla stanovena klasifikace
infekčnosti v tkáních pro ovce a kozy39);
klasifikace bude případně přezkoumávána
v návaznosti na nové vědecké důkazy.40)
1.2.10. Skot
Specifikované rizikové materiály uvedené v seznamu
příslušného nařízení Evropské unie41) se
považují za potenciálně vysoce TSE - infekční.
1.2.11. Deaktivace nebo odstranění přenosných původců
Výrobce
1.2.11.1. provádí kontrolu původu materiálu
u zdravotnických prostředků, v nichž
nemůže proběhnout proces
deaktivace/eliminace, aniž by u nich
došlo k nepřijatelné degradaci,
1.2.11.2. prohlašuje a podává důkazy v příslušné
dokumentaci, že výrobními procesy lze
odstranit nebo deaktivovat přenosné
původce, a to na základě prohlášení.
K odůvodnění deaktivačních/eliminačních
faktorů deaktivace či eliminace mohou být
použity důležité informace získané na
základě rešerše a analýzy příslušné
vědecké literatury, pokud jsou specifické
procesy uvedené v literatuře srovnatelné
s těmi, které jsou použity pro daný
prostředek. Tyto rešerše a analýzy by
měly zahrnout také dostupná vědecká
stanoviska případně přijatá Vědeckým
výborem EU. Tato stanoviska se považují
za reference v případech, kdy existují
rozdílné názory,
1.2.11.3. vytváří specifickou vědeckou deaktivační,
popřípadě eliminační studii, pokud
z rešerše odborné literatury nevyplyne
dostatečné odůvodnění, přičemž musí
zohlednit
1.2.11.3.1. identifikované nebezpečí
související s tkání,
1.2.11.3.2. identifikaci příslušných
modelových činitelů,
1.2.11.3.3. odůvodnění výběru
jednotlivých kombinací
modelových činitelů,
1.2.11.3.4. identifikaci etapy zvolené
k odstranění, popřípadě
deaktivaci přenosných
původců,
1.2.11.3.5. výpočet redukčních faktorů.
V konečné zprávě musí být uvedeny výrobní parametry
a limity kritické pro účinnost procesů deaktivace
nebo eliminace.
Používají se dokumentované postupy za účelem
zajištění, že se během rutinní výroby
zdravotnických prostředků užívají validované
výrobní parametry.
1.2.12. Množství výchozích tkání nebo derivátů zvířecího
původu nezbytných k výrobě jedné jednotky
zdravotnického prostředku
Výrobce
1.2.12.1. zhodnotí, jaké množství surových tkání
nebo derivátů je nezbytné pro výrobu
jedné jednotky zdravotnického prostředku,
1.2.12.2. posoudí, pokud je součástí výroby
zdravotnického prostředku purifikační
proces, zda může případně vést ke
koncentraci přenosných původců přítomných
ve výchozích tkáních nebo derivátech
zvířecího původu.
1.2.13. Tkáně nebo deriváty zvířecího původu, které
přicházejí do styku s uživateli
Výrobce
1.2.13.1. zohledňuje
1.2.13.1.1. množství tkání nebo derivátů
zvířecího původu,
1.2.13.1.2. kontaktní plochu, její
povrch, typ (například kůže,
sliznice, mozek) a stav
(například zdravá, nebo
poškozená),
1.2.13.1.3. typ tkání nebo derivátů
přicházejících do styku
s uživateli, a
1.2.13.1.4. předpokládanou dobu styku
zdravotnického prostředku
s tělem (včetně
bioresorpčního účinku),
1.2.13.2. bere v úvahu počet zdravotnických
prostředků, které mohou být použity
v daném postupu.
1.2.14. Cesta podání
Výrobce bere v úvahu cestu podání doporučenou
v informaci o výrobku, od nejvyššího rizika
k nejnižšímu.
1.3. Přezkoumání posouzení
Výrobce
1.3.1. zavádí a udržuje systematický postup pro
přezkoumání informací získaných o zdravotnickém
prostředku nebo podobných prostředcích;
v povýrobní fázi tyto informace hodnotí za účelem
stanovení jejich případné důležitosti pro
bezpečnost zdravotnického prostředku, zejména pokud je
1.3.1.1. odhaleno dříve nezjištěné nebezpečí,
1.3.1.2. předpokládané riziko vycházející
z nějakého nebezpečí dále nepřijatelné,
1.3.1.3. původní posouzení z nějakého důvodu
neplatné.
Pokud platí některý z výše uvedených bodů, bude na
výsledky hodnocení brán ohled v procesu řízení
rizik,
1.3.3. přezkoumá příslušná opatření řízení rizik pro
zdravotnický prostředek (včetně odůvodnění volby
tkáně nebo derivátu zvířecího původu),
1.3.4. znovu hodnotí a odůvodní účinnost dříve provedených
opatření kontroly rizik, pokud je možné, že se
změnilo zbytkové riziko nebo jeho přijatelnost;
výsledky tohoto hodnocení musí být zadokumentovány.
2.
HODNOCENÍ ZDRAVOTNICKÝCH PROSTŘEDKŮ TŘÍDY III NOTIFIKOVANÝMI
OSOBAMI
2.1. Výrobce
2.1.1. poskytuje notifikované osobě významné informace za
účelem zhodnocení aktuální strategie analýzy rizika
a řízení rizik,
2.1.2. zašle notifikované osobě jakoukoli novou informaci
o riziku TSE, kterou získá a která je pro zdravotnický
prostředek důležitá,
2.1.3. předkládá notifikované osobě k dodatečnému schválení
návrh jakékoli změny ve vztahu k procesu výběru,
odběru, manipulace a deaktivace, popřípadě eliminace,
která by mohla změnit výsledek dokumentací řízení
rizik, a to před jejím provedením.
2.2. Notifikovaná osoba
2.2.1. hodnotí aktuální strategii analýzy rizika a řízení
rizik na základě informací získaných od výrobce podle
bodu 2.1.,
2.2.2. dodatečně hodnotí, popřípadě schvaluje změny podle
bodu 2.1.3.
Příl.13
Formulář pro oznámení osoby nakládající se zdravotnickými prostředky
uvedené v § 13 odst. 1 nařízení vlády č. 336/2004 Sb.,
ve znění nařízení vlády č. 65./2011 Sb.

1) Používejte kódy zemí podle EN ISO 3166-1:2006, např.:
AT Rakousko,
BE Belgie,
BG Bulharsko,
CH Švýcarsko,
CY Kypr,
CZ Česká republika,
DE Německo,
DK Dánsko,
EE Estonsko,
ES Španělsko,
FI Finsko,
FR Francie,
GB Spojené království,
GR Řecko,
HU Maďarsko,
IE Irsko,
IS Island,
IT Itálie,
LI Lichtenštejnsko,
LT Litva,
LU Lucembursko,
LV Lotyšsko,
MT Malta,
NL Nizozemsko,
NO Norsko,
PL Polsko,
PT Portugalsko,
RO Rumunsko,
SE Švédsko,
SI Slovinsko,
SK Slovensko.
2) Vyplňte správný kód a název skupiny zdravotnického prostředku podle Globální nomenklatury
zdravotnických prostředků (GMDN).
2.
2.1. Formulář v elektronické podobě pro poskytnutí údajů podle bodu 1. a způsob jeho
vyplnění zveřejní Ministerstvo zdravotnictví na svých internetových stránkách.
2.2. Zplnomocněný zástupce podle § 3 písm. l) zákona o zdravotnických prostředcích
dále přikládá elektronickou kopii zmocnění výrobcem se sídlem mimo členské státy
v českém nebo v anglickém jazyce.
2.3. Pokud osoba zpracovatele formuláře není totožná s osobou oznamovatele, přikládá
tato osoba dále elektronickou kopii plné moci prokazující zmocnění k takovému úkonu.
Příl.14
Formulář pro oznámení osoby nakládající se zdravotnickými prostředky
uvedené v § 13 odst. 5 nařízení vlády č. 336/2004 Sb.,
ve znění nařízení vlády č. 65/2011 Sb.
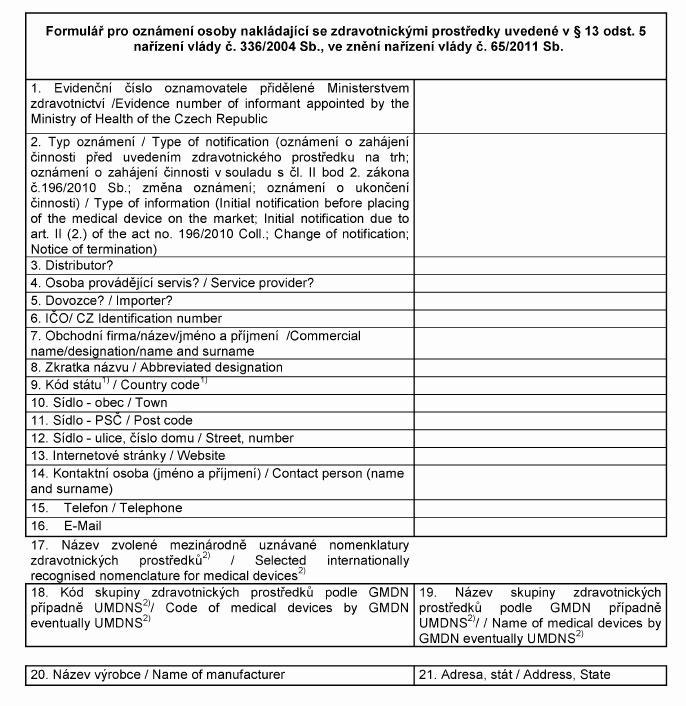
1) Používejte kódy zemí podle EN ISO 3166-1:2006, např.:
1) Please use the country codes according to EN ISO 3166-1:2006, e.g.:
AT Rakousko / Austria,
BE Belgie / Belgium,
BG Bulharsko / Bulgaria,
CH Švýcarsko / Switzerland,
CY Kypr / Cyprus,
CZ Česká republika / Czech Republic,
DE Německo / Germany,
DK Dánsko / Denmark,
EE Estonsko / Estonia,
ES Španělsko / Spain,
FI Finsko / Finland,
FR Francie / France,
GB Spojené království / United Kingdom,
GR Řecko / Greece,
HU Maďarsko / Hungary,
IE Irsko / Ireland,
IS Island / Iceland,
IT Itálie / Italy,
LI Lichtenštejnsko / Lichtenstein,
LT Litva / Lithuania,
LU Lucembursko / Luxembourg,
LV Lotyšsko / Latvia,
MT Malta / Malta,
NL Nizozemsko / Netherlands,
NO Norsko / Norway,
PL Polsko / Poland,
PT Portugalsko / Portugal,
RO Rumunsko / Romania,
SE Švédsko / Sweden,
SI Slovinsko / Slovenia,
SK Slovensko / Slovakia.
2) Jestliže má výrobce, případně zplnomocněný zástupce, distribuovaného zdravotnického
prostředku sídlo v jiném členském státě než České republice, který nevyžaduje údaje
podle Globální
nomenklatury zdravotnických prostředků (GMDN), je možno uvést údaje podle Univerzálního
nomenklaturního systému zdravotnických prostředků (UMDNS).
2) If the manufacturer or authorized representative of distributed medical device
resides in
other member state than Czech Republic, which does not require information by Global
Medical Device
Nomenclature (GMDN), the distributor can fill in information by Universal Medical
Device
Nomenclature System (UMDNS).
2.
2.1. Formulář v elektronické podobě pro poskytnutí údajů podle bodu 1. a způsob jeho
vyplnění
zveřejní Ministerstvo zdravotnictví na svých internetových stránkách.
2.2. Pokud osoba zpracovatele formuláře není totožná s osobou oznamovatele, přikládá
tato osoba
dále elektronickou kopii plné moci prokazující zmocnění k takovému úkonu.
Příl.15
Formulář pro oznámení zdravotnického prostředku
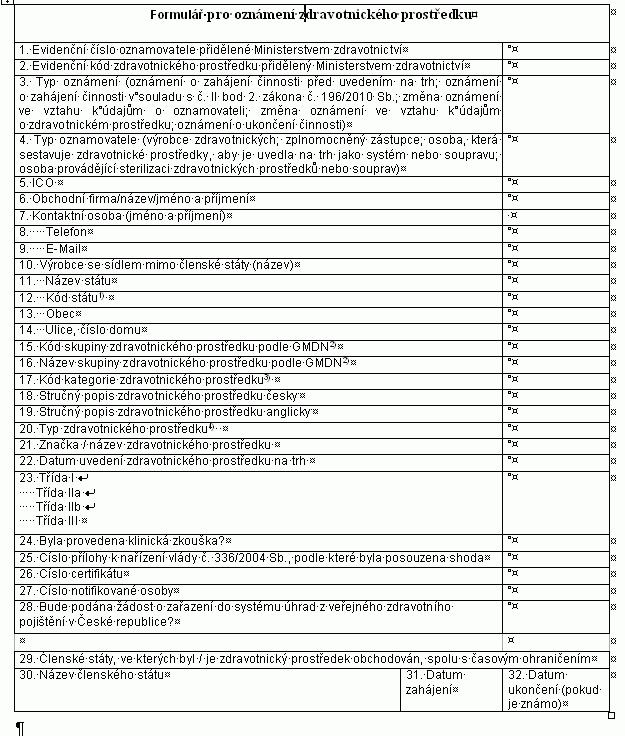
1) Používejte kódy zemí podle EN ISO 3166-1:2006, např.:
AT Rakousko,
BE Belgie,
BG Bulharsko,
CH Švýcarsko,
CY Kypr,
CZ Česká republika,
DE Německo,
DK Dánsko,
EE Estonsko,
ES Španělsko,
FI Finsko,
FR Francie,
GB Spojené království,
GR Řecko,
HU Maďarsko,
IE Irsko,
IS Island,
IT Itálie,
LI Lichtenštejnsko,
LT Litva,
LU Lucembursko,
LV Lotyšsko,
MT Malta,
NL Nizozemsko,
NO Norsko,
PL Polsko,
PT Portugalsko,
RO Rumunsko,
SE Švédsko,
SI Slovinsko,
SK Slovensko.
2) Vyplňte správný kód a název skupiny zdravotnického prostředku podle Globální nomenklatury
zdravotnických prostředků (GMDN).
3) ČSN EN ISO 15225:2010. Zdravotnický prostředek se označuje kódem první vhodné
kategorie, do které spadá.
4) Určitý výrobní model nebo varianta modelu podle specifikace uvedené v certifikátu.
2.
2.1. Formulář v elektronické podobě pro poskytnutí údajů podle bodu 1. a způsob jeho
vyplnění
zveřejní Ministerstvo zdravotnictví na svých internetových stránkách.
2.2. K formuláři se přikládá elektronická kopie závěrečné zprávy z klinického hodnocení,
certifikátu (byl-li vydán) a prohlášení o shodě v českém nebo anglickém jazyce.
2.3. K formuláři se přikládá elektronická kopie návodu k použití v českém jazyce.
2.4. Pokud osoba zpracovatele formuláře není totožná s osobou oznamovatele, přikládá
tato osoba
dále elektronickou kopii plné moci prokazující zmocnění k takovému úkonu.
Příl.16
Formulář o záměru provést klinické zkoušky
Clinical Investigation Notification Form
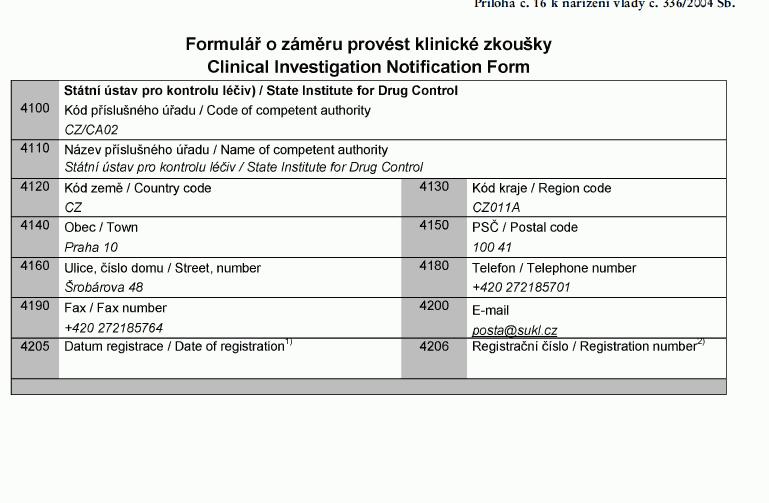
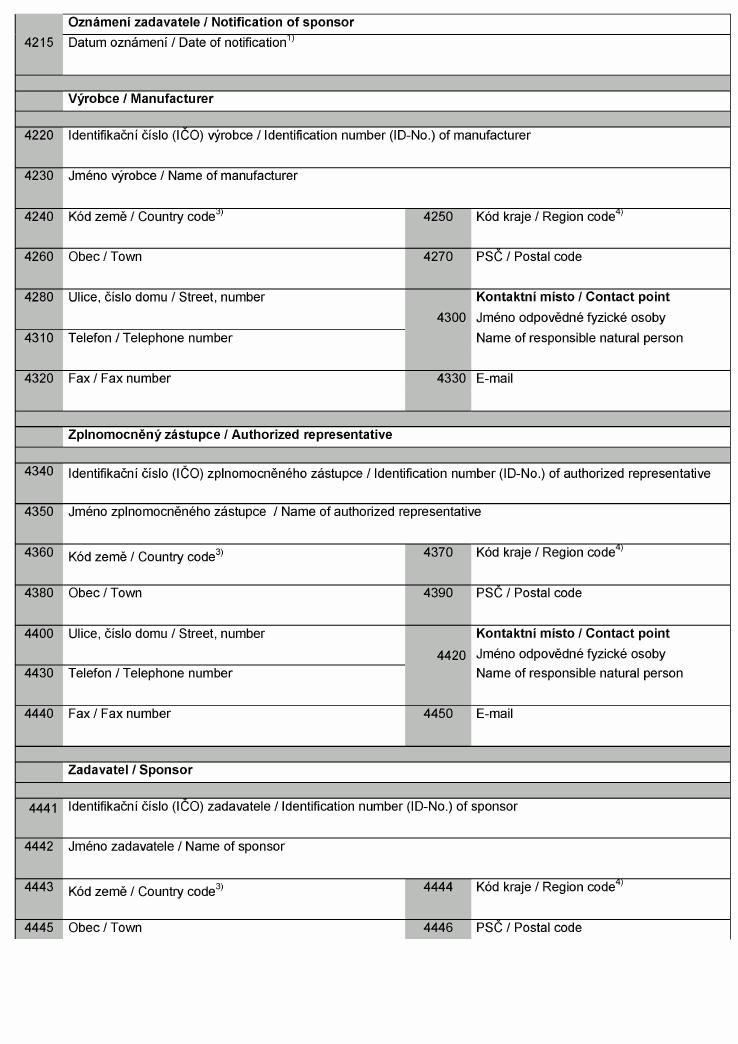

1) Používejte kódy zemí podle EN ISO 3166-1:2006, např.:
AT Rakousko,
BE Belgie,
BG Bulharsko,
CH Švýcarsko,
CY Kypr,
CZ Česká republika,
DE Německo,
DK Dánsko,
EE Estonsko,
ES Španělsko,
FI Finsko,
FR Francie,
GB Spojené království,
GR Řecko,
HU Maďarsko,
IE Irsko,
IS Island,
IT Itálie,
LI Lichtenštejnsko,
LT Litva,
LU Lucembursko,
LV Lotyšsko,
MT Malta,
NL Nizozemsko,
NO Norsko,
PL Polsko,
PT Portugalsko,
RO Rumunsko,
SE Švédsko,
SI Slovinsko,
SK Slovensko.
2) Vyplňte správný kód a název skupiny zdravotnického prostředku podle Globální nomenklatury
zdravotnických prostředků (GMDN).
3) ČSN EN ISO 15225:2010. Zdravotnický prostředek se označuje kódem první vhodné
kategorie, do které spadá.
4) Určitý výrobní model nebo varianta modelu podle specifikace uvedené v certifikátu.
2.
2.1. Formulář v elektronické podobě pro poskytnutí údajů podle bodu 1. a způsob jeho
vyplnění zveřejní Ministerstvo zdravotnictví na svých internetových stránkách.
2.2. K formuláři se přikládá elektronická kopie závěrečné zprávy z klinického hodnocení,
certifikátu (byl-li vydán) a prohlášení o shodě v českém nebo anglickém jazyce.
2.3. K formuláři se přikládá elektronická kopie návodu k použití v českém jazyce.
2.4. Pokud osoba zpracovatele formuláře není totožná s osobou oznamovatele, přikládá
tato osoba
dále elektronickou kopii plné moci prokazující zmocnění k takovému úkonu.
Vybraná ustanovení novel
Čl.II nařízení vlády č. 212/2007 Sb.
Přechodné ustanovení
1. Endoprotézy kyčle, endoprotézy kolena a endoprotézy ramena, u kterých
před 1. zářím 2007 bylo provedeno posouzení shody podle § 9 odst. 3
písm. a) nařízení vlády č. 336/2004 Sb., se podrobí doplňkovému posouzení
shody podle bodu 4 přílohy č. 2 k nařízení vlády č. 336/2004 Sb. do
1. září 2009. Tím není dotčena možnost předložit žádost o posouzení
shody podle § 9 odst. 1 písm. b) nařízení vlády č. 336/2004 Sb.
2. Endoprotézy kyčle, endoprotézy kolena a endoprotézy ramena, u kterých
před 1. zářím 2007 bylo provedeno posouzení shody podle § 9 odst. 3
písm. b) bodu 3 nařízení vlády č. 336/2004 Sb., se podrobí posouzení
shody jako zdravotnické prostředky třídy III podle § 9 odst. 1 písm.
b) bodu 1 nebo 2 nařízení vlády č. 336/2004 Sb. do 1. září 2010. Tím
není dotčena možnost předložit žádost o posouzení shody podle § 9 odst.
1 písm. a) nařízení vlády č. 336/2004 Sb.
3. Endoprotézy kyčle, endoprotézy kolena a endoprotézy ramena, u kterých
bylo provedeno posouzení shody podle § 9 odst. 3 písm. a) nařízení
vlády č. 336/2004 Sb. před 1. zářím 2007, lze uvádět na trh a do provozu
do 1. září 2009.
4. Endoprotézy kyčle, endoprotézy kolena a endoprotézy ramena, u kterých
bylo provedeno posouzení shody podle § 9 odst. 3 písm. b) bodu 3 nařízení
vlády č. 336/2004 Sb. před 1. zářím 2007, lze uvádět na trh do 1. září
2010; do provozu je lze uvádět i po tomto datu.
1) Směrnice Rady 93/42/EHS z 14. června 1993 o zdravotnických prostředcích,
ve znění směrnice Rady 98/79/ES z 27. října 1998, směrnice Evropského
parlamentu a Rady 2000/70/ES ze dne 16. listopadu 2000, směrnice Evropského
parlamentu a Rady 2001/104/ES ze dne 7. prosince 2001.
Směrnice Komise Evropských společenství 2003/12/ES ze dne 3. února 2003
o změně klasifikace prsních implantátů v rámci směrnice 93/42/EHS o
zdravotnických prostředcích.
Směrnice Komise Evropských společenství 2003/32/ES o zavedení podrobných
specifikací ohledně požadavků stanovených ve směrnici Rady č. 93/42/EHS,
pokud jde o zdravotnické prostředky vyráběné s využitím tkání zvířecího
původu ze dne 23. dubna 2003.
Nařízení Evropského parlamentu a Rady (ES) č. 1882/2003 ze dne 29. září
2003 o přizpůsobení ustanovení týkajících se výborů, které jsou nápomocny
Komisi při výkonu jejích prováděcích pravomocí, stanovených v právních
aktech Rady přijatých postupem podle článku 251 Smlouvy o ES, ustanovením
rozhodnutí 1999/468/ES.
Směrnice Komise 2005/50/ES ze dne 11. srpna 2005 o nové klasifikaci
endoprotéz kyčelního, kolenního a ramenního kloubu v rámci směrnice
Rady 93/42/EHS o zdravotnických prostředcích.
Směrnice Evropského parlamentu a Rady 2007/47/ES
ze dne 5. září 2007, kterou se mění směrnice
Rady 90/385/EHS o sbližování právních předpisů členských
států týkající se aktivních implantabilních zdravotnických
prostředků, směrnice Rady 93/42/EHS
o zdravotnických prostředích a směrnice 98/8/ES
o uvádění biocidních přípravků na trh.
2) § 2 zákona č. 123/2000
Sb., o zdravotnických prostředcích a o změně některých souvisejících
zákonů, ve znění zákona č. 130/2003 Sb. a zákona č. 274/2003 Sb.
3) § 2 odst. 2 písm. c) zákona č. 123/2000 Sb.
4) § 2 odst.
2 písm. f) zákona č. 123/2000 Sb., ve znění zákona č. 130/2003 Sb.
5) Zákon č. 378/2007 Sb., o léčivech a o změnách některých
souvisejících zákonů (zákon o léčivech), ve znění pozdějších
předpisů.
6) § 3 písm. f) zákona č. 123/2000 Sb., ve znění zákona
č. 130/2003 Sb.
7) Nařízení vlády č. 21/2003 Sb., kterým se stanoví technické
požadavky na osobní ochranné prostředky.
8) § 3 písm. e) zákona č. 123/2000 Sb., ve znění zákona č. 130/2003 Sb.
8a) Nařízení vlády č. 176/2008 Sb., o technických požadavcích
na strojní zařízení.
9) § 4a zákona
č. 22/1997 Sb., o technických požadavcích na výrobky a o změně a doplnění
některých zákonů, ve znění zákona č. 205/2002 Sb. a zákona č. 277/2003 Sb.
Čl. 5 bod 2 směrnice 93/42 EHS.
10) Nařízení
vlády č. 291/2000 Sb., kterým se stanoví grafická podoba označení CE.
11) § 3 písm.
a) zákona č. 123/2000 Sb., ve znění zákona č. 130/2003 Sb.
12) § 3 pím. l) zákona č. 123/2000 Sb., ve znění zákona č.
130/2003 Sb.
13) § 2 odst. 2 písm. e) zákona č. 123/2000 Sb., ve znění zákona č. 130/2003 Sb.
14) § 8 až 16 zákona č. 123/2000 Sb., ve znění zákona č. 130/2003 Sb.
15) § 2 odst. 2 písm. d) zákona
č. 123/2000 Sb.
16) Zákon č. 64/1986 Sb., o České obchodní
inspekci, ve znění zákona č. 240/1992 Sb., zákona č. 22/1997 Sb., zákona
č. 110/1997 Sb., zákona č. 189/1999 Sb., zákona č. 71/2000 Sb., zákona
č. 145/2000 Sb., zákona č. 102/2001 Sb., zákona č. 321/2001 Sb., zákona
č. 205/2002 Sb., zákona č. 309/2002 Sb. a zákona č. 226/2003 Sb.
17) § 14 zákona č. 22/1997 Sb., ve znění zákona č.
71/2000 Sb.
§ 36 odst. 1 písm. h) vyhlášky č. 307/2002 Sb., o radiační ochraně.
18) Nařízení vlády č. 18/2003 Sb., kterým se
stanoví technické požadavky na výrobky z hlediska jejich elektromagnetické
kompatibility.
19) § 6 odst. 1 zákona č. 64/1986 Sb., ve znění zákona č. 240/1992 Sb., zákona č.
22/1997 Sb. a zákona č. 145/2000 Sb.
20) § 19 odst. 1 zákona č. 22/1997
Sb., ve znění zákona č. 205/2002 Sb. a zákona č. 226/2003 Sb.
§ 7a odst. 1 zákona č. 64/1986 Sb., ve znění zákona č. 205/2002 Sb.
a zákona č. 226/2003 Sb.
21) Vyhláška č. 501/2000 Sb., kterou se stanoví formy, způsoby
ohlašování nežádoucích příhod zdravotnických prostředků, jejich evidování,
šetření a vyhodnocování, dokumentace a její uchovávání a následné sledování
s cílem předcházení vzniku nežádoucích příhod, zejména jejich opakování
(vyhláška o nežádoucích příhodách zdravotnických prostředků), ve znění
vyhlášky č. 304/2003 Sb.
22) § 3 písm. g) zákona č. 123/2000 Sb.
23) Například zákon
č. 18/1997 Sb., o mírovém využívání jaderné energie a ionizujícího
záření (atomový zákon) a o změně a doplnění některých zákonů, ve znění
zákona č. 83/1998 Sb., zákona č. 71/2000 Sb., zákona č. 132/2000 Sb.,
zákona č. 13/2002 Sb., zákona č. 310/2002 Sb. a zákona č. 320/2002 Sb.
24) § 7 odst. 7. písm. b) a § 11 odst. 2 a 4 zákona č. 22/1997
Sb., ve znění zákona č. 205/2002 Sb.
25) § 7 zákona č. 22/1997 Sb., ve znění zákona č. 205/2002 Sb.
25a) Zákon č. 356/2003 Sb., o chemických látkách a chemických
přípravcích a o změně a doplnění některých zákonů, ve
znění pozdějších předpisů.
26) Zákon č. 378/2007 Sb.
27) Zákon č. 505/1990 Sb., o metrologii, ve znění zákona č.
4/1993 Sb., zákona č. 20/1993 Sb., zákona č. 119/2000 Sb.,
zákona č. 13/2002 Sb., zákona č. 137/2002 Sb. a zákona č.
226/2003 Sb.
Vyhláška č. 264/2000 Sb., o základních měřících
jednotkách a ostatních jednotkách a o jejich označování.
28) Zákon č. 18/1997 Sb., ve znění zákona č. 83/1998 Sb., zákona
č. 71/2000 Sb., zákona č. 132/2000 Sb., zákona č. 13/2002 Sb.,
zákona č. 310/2002 Sb., zákona č. 320/2002 Sb. a zákona č.
279/2003 Sb.
29) § 2 odst. 2 písm. g) zákona č. 123/2000 Sb.
30) § 32 odst. 1 zákona č. 123/2000 Sb., ve znění zákona č.
130/2003 Sb.
30) § 3 písm. g) zákona č. 123/2000 Sb., ve znění pozdějších
předpisů.
31) Zákon č. 20/1966 Sb., o péči o zdraví lidu, ve znění zákona č.
210/1990 Sb., zákona č. 425/1990 Sb., zákona č. 548/1991 Sb.,
zákona č. 550/1991 Sb., zákona č. 590/1992 Sb., zákona č.
15/1993 Sb., zákona č. 161/1993 Sb., zákona č. 307/1993 Sb.,
zákona č. 60/1995 Sb., nálezu Úst. soudu publikovaného pod č.
206/1996 Sb., zákona č. 14/1997 Sb., zákona č. 79/1997 Sb.,
zákona č. 110/1997 Sb., zákona č. 83/1998 Sb., zákona č.
167/1998 Sb., zákona č. 71/2000 Sb., zákona č. 123/2000 Sb.,
zákona č. 132/2000 Sb., zákona č. 149/2000 Sb., zákona č.
258/2000 Sb., zákona č. 164/2001 Sb., zákona č. 260/2001 Sb.,
zákona č. 290/2002 Sb., zákona č. 285/2002 Sb., zákona č.
320/2002 Sb., zákona č. 130/2003 Sb., nálezu Úst. soudu
publikovaného pod č. 211/2003 Sb., zákona č. 274/2003 Sb.
a zákona č. 356/2003 Sb.
Zákon č. 160/1992 Sb., o zdravotní péči v nestátních
zdravotnických zařízeních, ve znění zákona č. 161/1993 Sb.,
zákona č. 258/2002 Sb., zákona č. 285/2002 Sb. a zákona č.
320/2002 Sb.
32) § 3 písm. d) zákona č. 123/2000 Sb.
33) § 11 odst. 3 zákona č. 22/1997 Sb., ve znění zákona č.
71/2000 Sb.
34) § 11 odst. 2 písm. e) zákona č. 22/1997 Sb.
35) § 3 písm. c) zákona č. 123/2000 Sb., ve znění zákona č.
130/2003 Sb.
36) Nařízení Evropského parlamentu a Rady (ES) č. 999/2001 ze dne
22. května 2001, o stanovení pravidel pro prevenci, tlumení
a eradikaci některých přenosných spongiformních encepalopatií.
37) Čl. 2.3.13.2. bodu 1 Mezinárodního kodexu zdraví zvířat
(Office International des Épizooites) - OIE; je dálkově
přístupný na adrese
www.oie.int/eng/normes/Mcode/A_00067.htm.
38) Nařízení Evropského parlamentu a Rady č. 1774/2002 ze dne 3.
října 2002, kterým se stanoví zdravotní pravidla týkající se
živočišných vedlejších produktů, které nejsou určeny k lidské
spotřebě.
39) Příloha stanoviska Vědeckého řídícího výboru ze dne 22. až
23. července 1999 (Policy of breeding and genotyping sheeps)
obsahuje tabulku, která byla aktualizována stanoviskem
Vědeckého řídícího výboru přijatém 10. - 11. ledna 2002 (TSE
infectivity distributed in ruminant tissues - state of
knowledge December 2001); je dálkově přístupná na adrese
http://europa.eu.int/comm/food/fs/sc/ssc/outcome_en.html.
40) Například za použití příslušných stanovisek vědeckých výborů,
výboru pro hromadně vyráběné léčivé přípravky a opatření
Komise upravujících použití materiálů představujících riziko
TSE. Přehled odkazů na příslušné dokumenty či stanoviska bude
zveřejněn v Úředním věstníku Evropské unie a po přijetí
příslušného rozhodnutí Komise bude vytvořen jejich seznam.
41) Nařízení ES č. 999/2001.